酢酸
| 酢酸 | |
|---|---|
IUPAC名 酢酸(許容慣用名) | |
| 識別情報 | |
CAS登録番号 | 64-19-7 |
E番号 | E260 (防腐剤) |
KEGG | C00033 |
SMILES
| |
| 特性 | |
化学式 | C2H4O2 |
モル質量 | 60.05 |
示性式 | CH3COOH |
| 外観 | 無色の液体 |
密度 | 1.049(液体) |
| 相対蒸気密度 | 2.1 |
融点 | 16.7 °C, 290 K, 62 °F |
沸点 | 118 °C, 391 K, 244 °F |
酸解離定数 pKa | 4.76 |
屈折率 (nD) | 1.3715 |
| 危険性 | |
NFPA 704 |  2 2 0 |
| 出典 | |
ICSC | |
| 特記なき場合、データは常温 (25 °C)・常圧 (100 kPa) におけるものである。 | |
酢酸(さくさん、醋酸、英: acetic acid)は、簡単なカルボン酸の1種である。IUPAC命名法では、酢酸は許容慣用名であり、系統名はエタン酸 (ethanoic acid) である。純粋なものは冬に凍結することから氷酢酸(ひょうさくさん)と呼ばれる。2分子の酢酸が脱水縮合すると別の化合物の無水酢酸となる。
食酢(ヴィネガー)に含まれる弱酸で、強い酸味と刺激臭を持つ。遊離酸・塩・エステルの形で植物界に広く分布する。酸敗したミルク・チーズのなかにも存在する。
試薬や工業品として重要であり、合成樹脂のアセチルセルロースや接着剤のポリ酢酸ビニルなどの製造に使われる。全世界での消費量は年間およそ6.5メガトンである。このうち1.5メガトンが再利用されており、残りは石油化学原料から製造される[1]。生物資源からの製造も研究されているが、大規模なものには至っていない。
目次
1 歴史
1.1 酢の利用
1.2 発見と研究
1.3 工業生産
2 名称
3 性質
3.1 物理的性質
3.2 二量体
3.3 酸性度
4 化学反応
4.1 酸としての反応
4.2 カルボキシル基の変換
4.3 脱水
4.4 メチル基での反応
5 生化学
6 製造
6.1 メタノールのカルボニル化
6.2 アセトアルデヒドの酸化
6.3 アルカンの酸化
6.4 エチレンの酸化
6.5 発酵
6.5.1 酸化的発酵
6.5.2 無気性発酵
7 用途
7.1 酢酸ビニルモノマー
7.2 無水酢酸
7.3 エステルの製造
7.4 酢
7.5 溶媒
7.6 その他
8 出典
9 参考文献
10 関連項目
歴史

バルサミコ酢と赤・白のワインビネガー
酢の利用
酢の歴史は文明と同程度に古く、酢酸産生菌はいたるところに存在する。そして、ビールやワインなど酒を醸造する文明は、酒を大気にさらすと、自然に酢ができることを発見することになる[2]。紀元前、ギリシャの哲学者テオプラストスやローマのウィトルウィウス、プリニウスは酢が金属に作用して芸術に有用な顔料、たとえば鉛白(塩基性炭酸鉛)やビリジリス(酢酸銅(II) を含む銅塩の緑色の混合物)となることについて著述している[3][4]。また、酢はその時代にはローマでは治療の目的[2]、エジプトでは死体の保存[5]に用いられていたともされている。古代ローマ人は酸っぱくなったワインを鉛の壷で煮沸すると、サパあるいはデフルタムと呼ばれる非常に甘いシロップができることを見出している。サパやデフルタムの甘さは含まれる酢酸鉛(II) によるもので、その物質は鉛糖 (sugar of lead) とか土の糖 (sugar of saturn) と呼ばれ好まれたが、ローマ貴族の間で鉛中毒を引き起こした[6]。
発見と研究
8世紀にジャービル・イブン=ハイヤーン(ゲベル)は初めて酢の蒸留によって酢酸を得ている[7]。またルネサンス時代には、錬金術師たちは酢酸銅(II) などの金属酢酸塩を乾留して氷酢酸を製造した[8]。最初にそのような製法で酢酸を作り出したのはバシル・バレンティンとされている[2]。16世紀のドイツの化学者アンドレアス・リバヴィウスは、氷酢酸の製法と、得られた氷酢酸と酢との物性の比較について著述している[8]。そのように、酢の中には水が存在するため物性が酢酸と異なることから、氷酢酸と酢の中の酸は別の物質であると長く信じられていたが[9]、18世紀になるとフランス人化学者のピエール・エディにより両者が同一であることが示された[10][11]。
1845年にドイツ人化学者のヘルマン・コルベは無機物から有機物である酢酸を合成できることを示した[12]。その反応は、まず二硫化炭素から四塩化炭素への塩素化で始まり、次いでテトラクロロエチレンへの熱分解、そしてトリクロロ酢酸への水性塩素化、最後に電解還元による酢酸の生成、というものだった[13]。この結果はフリードリヒ・ウェーラーの尿素合成による生気論の否定を決定付けた[14]。一方ルイ・パスツールは1862年に酢酸菌を発見し、酢の醸造に利用されるようになったが、得られる濃度が低いため工業用の酢酸の製造には適していなかった[15]。
工業生産

酢酸の精製・濃縮工場(1884年)
1910年頃までは、氷酢酸は大部分が木材の乾留で得られる木酢液から作られていた[16]。イギリスでは1820年ごろから[17]、日本では明治時代に[15]、この方法による酢酸の製造が始められていた。木酢液を水酸化カルシウム(石灰乳)で処理して生成する酢酸カルシウムを硫酸で酸性化することにより、酢酸が分離される[16]。1917年頃のドイツは年間およそ1万トンの氷酢酸を生産していたが、その30%はインディゴの製造に充てられていた[16]。1910年代の半ば以降からは、ドイツとカナダでカルシウムカーバイドから得られるアセチレンを原料とした酢酸の製造が始められた[18]。カルシウムカーバイドはコークス(石炭の乾留物)を酸化カルシウム(生石灰)とともに電気炉で加熱することにより得られるが、ドイツは石油に乏しいが石炭を産出すること、カナダは水力発電による電力を有することが有利な点であった[19]。日本でも水力発電の発達に伴い、1928年以降この製法で酢酸が作られるようになった[20][21]。1937年に日本窒素肥料(現チッソ)も同法による酢酸の製造を開始したが、アセチレンの酸化に用いられる硫酸水銀(II) がのちに水俣病の原因となった[22]。
やがて石油化学工業が発展すると、酢酸の製造法はエチレンやアルカンを原料とするものに変わっていった[23]。さらに1960年代にドイツのBASFによってコバルト触媒、1970年にアメリカ合衆国のモンサントによってロジウム触媒を用いたメタノールのカルボニル化反応が開発・実用化され、それ以降はこれらが工業的に主要な氷酢酸の製造法となった[24]。
名称

凍った酢酸(氷酢酸)
日本語の「酢酸」は江戸時代後期に宇田川榕菴が著書舎密開宗で用いたのが最初である[25]。オランダ語 azijnzuur の訳語であり、これはさらにドイツ語 Essigsäure、英語 acetic acid の訳語であった。これらの名称はそのまま現代でも使われ、acetic acid や「酢酸」はIUPAC命名法における許容慣用名[26]かつ優先IUPAC名 (PIN)[27] およびその訳語である。IUPAC系統名は「エタン酸」ethanoic acid であり[28]、これは母体化合物「エタン」 ethane にカルボン酸官能基を表す接尾辞「酸」 -oic acid を付加したものである。
有機化学ではアセチル基 CH3C(=O)− の略号 Ac を用いて文章や化学式中で AcOH または HOAc と略記される。酢酸のエステルや塩は英語ではアセテート(アセタート) acetate と呼ばれる。たとえばエチルエステルの酢酸エチルは ethyl acetate、アンモニウム塩の酢酸アンモニウムは ammonium acetate である。
純粋な酢酸は、融点が約摂氏16度であることから、温度がそれを下回ると固体になり、特にその外見が氷に似ていることから「氷酢酸」(glacial acetic acid) とも呼ばれる[29]。このため、日本程度の気候であっても、地方にもよるが特に冬期には室温で固体になることが珍しくない物質のひとつでもある。
また酢酸は、古くは単に vinegar (酢)、 酢の蒸留によって得られたことから acetous acid (酢の酸)、木材の乾留で得られることから pyroligneous acid (火木酸)、ほか spilit of verdigris (ビリジリスの精)や wood vinegar (木酢)とも呼ばれた[5]。日本でも
英語 acetic acid の語源は酢を意味するラテン語 acetum と「鋭い」を意味する acer に由来する[30][31]。ここから派生して「アセト」acet(o)- の語は酢酸から得られたり構造が類似する化合物などにも用いられる。たとえばアセトン、アセトニトリル、アセトイン、アセトフェノン、アセチル基がそうである[31]。また炭素原子の数が同じく2個であるビニル基(ビニルラジカル)も古くは acetic acid を語源としてアセチルラジカル acetyl radical と呼ばれており[31]、これに由来する名称を持つ化合物としてアセチレンやアセナフテンなどがある[31][32]。
性質
物理的性質
| 濃度 (重量%) | 比重 (25 °C/4 °C) |
|---|---|
| 100 | 1.0553 |
| 90 | 1.0713 |
| 80 | 1.0748 |
| 70 | 1.0733 |
| 60 | 1.0685 |
| 50 | 1.0615 |
| 40 | 1.0523 |
純粋な酢酸は、直鎖状の飽和炭化水素鎖を持ったカルボン酸の中では比重が高く、1を超えている。常温・常圧において酢酸よりも炭素数の多いプロピオン酸(プロパン酸)などは概ね比重が1を下回っており、酢酸よりも比重が大きいのは酢酸よりも炭素数が少ない蟻酸である。また、常温常圧において酸味と刺激臭を持つ無色透明の液体である。常圧における融点は約16.7 ℃、沸点は約118 ℃である。なお、このうち融点は低分子の直鎖状の飽和炭化水素鎖を持ったカルボン酸としては高く、酢酸よりも炭素鎖の長いプロパン酸、酪酸(ブタン酸)、吉草酸(ペンタン酸)、カプロン酸(ヘキサン酸)、エナント酸(ヘプタン酸)の融点よりも高い。常圧において炭化水素鎖2つの酢酸とほぼ同じ融点を持つのは、炭化水素鎖8つのカプリル酸(オクタン酸)である。しかし、酢酸の場合は少量の水と混合すると融点が大きく低下し[2]、水の割合が約40 %の時に最低値-26.75 ℃となる[29]。酢酸と水との混合液を冷却した時、これよりも水が少ないと酢酸が、多いと氷が晶出する[29]。酢酸と水との混合液を加熱しても、水との共沸は起こらない[33]。また、水との混合により比重が増加し、酢酸の濃度が約80%のとき最も大きくなり[16]、43%のとき純粋な酢酸と同じになる[29]。蒸気を燃やすとき、炎は淡青色である[29]。
酢酸は水、アセトニトリル、エタノール、酢酸エチル、クロロホルム、ベンゼン、エーテル、石油エーテルと任意の割合で混和する[34][35]。オクタンなど長鎖炭化水素には溶けにくく、溶解度は鎖が長くなるほど低くなる[36]。二硫化炭素には不溶である[35]。比誘電率は約6であり、あまり高くはないが[37]、無機塩や糖といった極性化合物を溶かすことができる[38]。また単体硫黄 S8、ヨウ素 I2 など無極性の分子も酢酸に溶ける[38]。ほかにゼラチン、フィブリン、アルブミン、樟脳、ニトロセルロースも溶ける[2]。酢酸の純度を知る古い方法としてレモン油を加えるというものがあり、これは純粋な酢酸であれば重量で10%のレモン油を完全に溶かすことによる[2]。
酢酸を構成する炭素原子と酸素原子は平面上に位置し、結合角は C−C=O と C−C−OH が119°、O=C−OH が122°で、結合距離は C−C が 152 pm、C=O が125 pm、C−OH が131 pm である[39]。
二量体

酢酸の二量体。破線は水素結合を示す。
酢酸は水素結合を介して2分子が結合した、環状の二量体を形成する[39]。気体状態では電子回折により[40]、固体状態ではX線結晶構造解析により[41]、それぞれ構造が確認されている。純粋な液体状態ではほとんど単量体としては存在しないが、二量体となっているか、もしくは直鎖状あるいは環状の多量体となっているとされる[42]。希薄な溶液の場合、四塩化炭素[43]やベンゼンなどの非プロトン性溶媒中では二量体を形成するが、水などプロトン性の溶媒中では単量体として存在する[44][45]。
この二量体を形成するという性質のため、酢酸(分子量60.05)の沸点は水素結合を作らない酢酸メチル(分子量74.08、沸点 57 °C[46])よりも高く、分子量が2倍程度のオクタン(分子量114.23、沸点 125 °C)に近い[47]。
酸性度
酢酸のカルボキシル基 −COOH{displaystyle {ce {-COOH}}}
- CH3CO2H+H2O⟶CH3CO2−+H3O+{displaystyle {ce {CH3CO2H + H2O -> CH3CO2^- + H3O^+}}}


共鳴構造も加味した、実際の酢酸の解離。プロトンを放出したカルボニル基は、炭素と酸素全体に負電荷が分散する。上記の図では、破線で書かれている部分に負電荷が広がっている。このため、比較器安定なアニオンとして存在できる。
この性質のため、酢酸は酸性を持つ。酢酸は弱酸であり、水溶液中でのpKaはおよそ4.76である[49]。すなわち、1.0 mol/L の水溶液のpHは2.4となり、全体の0.4%が解離していることになる[50]。酢酸は塩酸や硫酸などの無機酸よりは弱く、炭酸やフェノール、アルコールよりは強い酸である[48][49]。
なお、酢酸の2位の炭素に結合する水素が、フッ素や塩素や臭素やヨウ素に置換されると酸性度が上がることが知られている[51]。特にトリフルオロ酢酸やトリクロロ酢酸は強酸として知られる。
化学反応
酸としての反応
塩基である炭酸カリウムと混合すると、中和により酢酸カリウムが生成する。これを単離し酢酸に溶かして加熱すると脱水して二酢酸カリウムとなり、200 °C 以上でさらに反応して無水酢酸と酢酸カリウムに分離する[52]。
- CH3COOH+K2CO3⟶CH3COOK+H2CO3{displaystyle {ce {CH3COOH + K2CO3 -> CH3COOK + H2CO3}}}
- CH3COOH+CH3COOK⟶(CH3COO)2KH+H2O{displaystyle {ce {CH3COOH + CH3COOK -> (CH3COO)2KH + H2O}}}
- (CH3COO)2KH⟶(CH3CO)2O+CH3COOK{displaystyle {ce {(CH3COO)2KH -> (CH3CO)2O + CH3COOK}}}



酢酸はアルミニウム、銅、銀、チタン、ジルコニウムを腐食しないので、これらの金属は酢酸の容器として利用できる。一方、鉛やステンレスは酢酸によって侵される[53]。これらのうち鉛の場合は酢酸鉛となって鉛が大量に溶出してくる恐れがあり、これを摂取すると鉛中毒の原因となり得るため危険である。また、酢酸はマグネシウムと反応して水素と酢酸マグネシウムを生じる[54]。
- 2CH3COOH +Mg⟶(CH3COO)2Mg +H2{displaystyle {ce {2 CH3COOH + Mg -> (CH3COO)2Mg + H2}}}

カルボキシル基の変換


酢酸はカルボン酸として一般的な反応性を示す。たとえば硫酸を触媒としてアルコールと共に加熱すると酢酸エステルが生成する。これはフィッシャーエステル合成反応と呼ばれる方法である。可逆反応(平衡反応)であるため、エステル生成物を効率よく得るには出発物質を過剰に使用する必要があり、イソペンチルアルコールとの反応による酢酸イソペンチルの合成では、過剰量の酢酸が用いられる[55]。
- CH3COOH +C5H11OH⟶CH3COOC5H11 +H2O{displaystyle {ce {CH3COOH + C5H11OH -> CH3COOC5H11 + H2O}}}

酢酸からのエステル合成法としては他にアルケンへの付加があり、ヘテロポリ酸を触媒としてエチレンから酢酸エチルが得られる[56]。
- :CH3COOH+CH2=CH2⟶CH3COOCH2CH3{displaystyle {ce {:CH3COOH + CH2=CH2 -> CH3COOCH2CH3}}}


アセトアミド(左)と塩化アセチル(右)の分子模型
炭酸アンモニウムと混合して加熱すると、酢酸アンモニウムの生成と脱水を経てアセトアミドが得られる。この反応は蒸留によって酢酸を除きながら行い、さらに沸点のより高い残渣を引き続いて蒸留し、純粋な目的物を得る[57]。アンモニアを使っても同様な反応が起きる[57]。
- 2CH3COOH+(NH4)2CO3⟶2CH3COONH4+H2CO3{displaystyle {ce {2 CH3COOH + (NH4)2CO3 -> 2 CH3COONH4 + H2CO3}}}
- CH3COONH4⟶CH3CONH2+H2O{displaystyle {ce {CH3COONH4 -> CH3CONH2 + H2O}}}


カルボン酸塩化物である塩化アセチルは、酢酸と三塩化リンや塩化チオニルなどの反応で得られる[58]。塩化チオニルは過剰量を用いるが、蒸留では塩化アセチルと分離しづらいため、余ったぶんは蟻酸と反応させて分解する[58]。
- CH3COOH+SOCl2⟶CH3COCl+SO2+HCl{displaystyle {ce {CH3COOH + SOCl2 -> CH3COCl + SO2 + HCl}}}

脱水

無水酢酸(左)とケテン(右)の分子模型
加熱により2分子間で脱水縮合を起こし、無水酢酸を与える。環状の酸無水物を生成する場合を除き、このような反応はほかのカルボン酸では起こらない[59]。
2CH3COOH⟶(CH3CO)2O +H2O{displaystyle {ce {2 CH3COOH -> (CH3CO)2O + H2O}}}(800 °C)
また、リン酸エステルの存在下に 700–800 °C に加熱すると、分子内脱水によりケテン(エテノン)を生じる[60]。
CH3COOH⟶CH2=C=O+H2O(O=P(OCH2CH3)3){displaystyle {ce {CH3COOH -> CH2=C=O + H2O (O=P(OCH2CH3)3)}}}(700–800 °C)
さらに、酢酸はケテンに付加して無水酢酸を与える[61]。
- CH3COOH+CH2=C=O⟶(CH3CO)2O{displaystyle {ce {CH3COOH + CH2=C=O -> (CH3CO)2O}}}

メチル基での反応

クロロ酢酸の分子模型
日光を当てながら酢酸と塩素を反応させると、水素原子と塩素原子が交換したクロロ酢酸が生成する[62]。この反応はラジカルの発生を含む機構で進行し、ジクロロ酢酸やトリクロロ酢酸が副生成物となるが、触媒の使用によりそれらの生成を抑えることもできる[63]。
- CH3COOH+Cl2⟶CH2ClCOOH+HCl{displaystyle {ce {CH3COOH + Cl2 -> CH2ClCOOH + HCl}}}

同様にして臭素とリン触媒を使って酢酸からブロモ酢酸を作ることができる[64]。この合成法はヘル・ボルハルト・ゼリンスキー反応と呼ばれる。
生化学
酢酸は生体内で活性化体であるアセチルCoA(アセチル補酵素A)としてさまざまな役割を果たす。アセチルCoAは活性酢酸とも呼ばれる[65]酢酸のチオエステル体であり、CoASHはよい脱離基として働くため酢酸そのものよりも置換反応が起こりやすい[66]。

アセチルCoAの分子模型。左端の黄色い硫黄原子上にアセチル基が結合している。
アセチルCoAは体内での代謝経路、すなわち、解糖系による糖からのピルビン酸の生成とそれに続く補酵素Aとの結合[67]、脂肪酸のβ酸化の繰り返しによる逐次分解[68]、そしてアミノ基転移を経るアミノ酸の異化[69]によって生成する。また、アセチルCoAリガーゼ(アセチルCoA合成酵素)により酢酸と補酵素Aから直接合成される。2種類のアセチルCoAリガーゼにより以下の反応が起こる。
酢酸 + ATP + CoA → アセチルCoA + AMP + 二リン酸
アセチルCoAリガーゼ (EC 6.2.1.1)[70]
酢酸 + ATP + CoA → アセチルCoA + ADP + リン酸
酢酸CoAリガーゼ (ADP生成) (EC 6.2.1.13)[71]
生成したアセチルCoAはクエン酸回路でのエネルギー生産や、脂肪酸の合成、メバロン酸経路によるテルペノイド・ステロイドの生合成などに利用される[65][72][73]。クエン酸回路による代謝では、酢酸は最終的に二酸化炭素と水になる[74]。
アセチルコリンの分子模型
アセチルコリンはコリンとアセチルCoAとから合成される神経伝達物質であり、神経細胞の末端において小胞体に蓄えられる。刺激を受けると放出され、受容体に結合することによって信号を伝達する。役目を終えるとすぐにアセチルコリン加水分解酵素によってコリンと酢酸とに分解される[75]。
また、酒や酒を含む食品を摂取すると人体では酢酸が生産される。エタノールはアセトアルデヒドを経て酵素アルデヒドデヒドロゲナーゼにより酢酸に変換される(「エタノールと人体」も参照)。
メタン生成古細菌(メタン菌)と呼ばれる古細菌のうち、メタノトリクス属(メタノサエタ属)やメタノサルキナ属は酢酸を代謝してメタンを生成することが知られており、汚水処理やバイオマス生産へ利用されている[76][77]。
サソリモドキというクモ綱の節足動物は、後腹部から酢酸を噴射して身を守るとされている[78]。
製造
化学合成とバクテリアによる発酵の両方によって作られる。今日では発酵法は全世界での生産量の10%を占めるに過ぎないが、食品の品質に関する法律は食用の酢として用いられる場合に生物由来であることを求めるものが多いため[79]、依然として食酢の製造には重要である[80]。化学工業で用いられる酢酸のおよそ8割はメタノールのカルボニル化によって作られている[81]。
全世界での酢酸の純生産量はおよそ年5メガトンと見積もられ、その半分はアメリカ合衆国によるものである。ヨーロッパでの生産量は年に約1メガトンだが減少傾向にあり、日本では年0.7メガトンである(酢酸の2008年度日本国内生産量は500,211トン、消費量は181,799トンである[82])。残り1.5メガトンは毎年再利用されており、都合、全世界での市場流通量は年6.5メガトンとなる[83][84]。
メタノールのカルボニル化
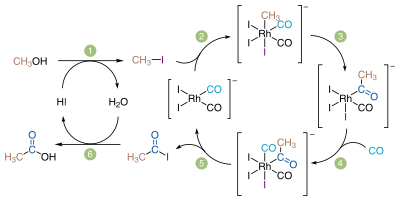
モンサント法における触媒サイクル
大部分の酢酸はこの方法によって生産されている。メタノールと一酸化炭素を下記の反応式に従って反応させる[85]。
- CH3OH+CO⟶CH3COOH{displaystyle {ce {CH3OH + CO -> CH3COOH}}}

この方法は中間体としてヨードメタンを含む3段階の過程である。2段階目の反応は触媒を必要とし、通常これには第9族元素の金属錯体が用いられる。
- CH3OH+HI⟶CH3I+H2O{displaystyle {ce {CH3OH + HI -> CH3I + H2O}}}
- CH3I+CO⟶CH3COI{displaystyle {ce {CH3I + CO -> CH3COI}}}
- CH3COI+H2O⟶CH3COOH+HI{displaystyle {ce {CH3COI + H2O -> CH3COOH + HI}}}



メタノールと一酸化炭素は共に簡単に得られる原料であるため、メタノールのカルボニル化は長らく酢酸製造の魅力的な方法であった。セラニーズ社のヘンリー・ドレフュス (Henry Drefyus) は本法の試験プラントを1925年頃に既に開発していた[86]。しかし、腐食性の混合物を200気圧という高圧下で反応させることができる装置の材料が金やグラファイトのほかになかったため、当時は工業化することができなかった[87]。最初の工業化はコバルト触媒を用いる方法で、ドイツの化学会社 BASF社によって1960年に小型プラントが開発された[85]。材質の問題はハステロイの登場により解決されている[85][87]。1968年にロジウム触媒 (cis-[Rh(CO)2I2]−) が発見され、より低圧でほとんど副生物を発生させずに反応を進行させることが可能になった。この触媒を使用した最初のプラントは1970年にアメリカの化学会社モンサント社によって建設され、ロジウム触媒によるメタノールのカルボニル化が酢酸製造の主要な方法になった(モンサント法)[88]。1990年代後期、化学会社BPケミカルズ社がロジウムをイリジウムで置き換えたカティバ触媒 ([Ir(CO)2I2]−) を開発した[89]。この触媒はよりグリーン・高効率であり[90]、同じプラントで利用できるモンサント法にとって代わった。
アセトアルデヒドの酸化
モンサント法が工業化される以前には、大部分の酢酸はアセトアルデヒドの酸化によって製造されていた。メタノールのカルボニル化と競合するほどではないが、依然として第2の重要な製造法である。アセトアルデヒドはブタンや軽ナフサの酸化[91]、あるいはエチレンの酸化(ワッカー法)によって作られる[92]。
酢酸コバルトや酢酸マンガンを触媒とした、アセトアルデヒドの空気酸化によって酢酸が得られる[93]。
- 2CH3CHO +O2⟶2CH3COOH{displaystyle {ce {2 CH3CHO + O2 -> 2 CH3COOH}}}

反応は過酢酸の生成を経るが、条件を調整することにより、これを主生成物とすることもできる。副生成物として二酸化炭素、メタノール、酢酸メチル、蟻酸、蟻酸メチル、ホルムアルデヒドが含まれるが、蒸留により精製される[94]。
アルカンの酸化
ブタンや軽ナフサを空気中でマンガン、コバルト、クロムなどの金属イオンの存在下に加熱すると、ヒドロペルオキシドが生成したのちに分解し、酢酸を与える[91]。
- 2C4H10+5O2⟶4CH3COOH+2H2O{displaystyle {ce {2 C4H10 + 5 O2 -> 4 CH3COOH + 2 H2O}}}

一般的に、ブタンが液体状態である限界の高温で反応を進行させられるように温度と圧力を設定する。典型的には 160–200 °C、4–8メガパスカルである。メチルエチルケトン、酢酸エチル、蟻酸、プロパン酸などが副生物として得られる[95]。これらの副生物も市場価値があるため、分離の手間も含めて充分に採算が取れれば、これらがより多く生成するように条件が変更されることもある。
エチレンの酸化
アセトアルデヒドはワッカー法によりエチレンから作ることができ、これを上記の方法で酸化する。より安価な1段階のエチレンからの酢酸の製造法が昭和電工によって工業化され、1997年に大分県でエチレン酸化プラントが開業された[96]。その方法ではタングストケイ酸などのヘテロポリ酸上に担持されたパラジウム触媒を用いる[97]。エチレンの価格によっては、小さめのプラント(100–150キロトン/年)でメタノールのカルボニル化と競合する。しかしながら2009年に昭和電工は大分での酢酸製造設備を停止し、メタノール法の酢酸をマレーシア、中国などから輸入するようになった。これはナフサ価格上昇にともない、エチレン価格も高騰したため、メタノール法酢酸製造プラントに対抗できなくなったためである[98]。これにより現在日本で酢酸を製造するプラントは協同酢酸の1社のみとなった。
発酵
酸化的発酵
人類の歴史の大部分において、酢酸は酢の形でアセトバクター属 (Acetobacter) の細菌によって作られてきた。充分な量の酸素がある環境、すなわち好気的な条件において、それらのバクテリアはエタノールを含有する様々な食品から食酢(以下「酢」とのみ表記)を作り出す。普通に使われるのはリンゴ酒、ワイン、発酵させた穀物、麦芽、米、すりつぶしたジャガイモである。バクテリアによって促進される化学反応は、全体として以下のようなものである。
- C2H5OH+O2⟶CH3COOH+H2O{displaystyle {ce {C2H5OH + O2 -> CH3COOH + H2O}}}

すなわちエタノールが持つ水酸基の酸化を行っているのである。アセトバクター属を接種して保温すると、空気に触れている部分が数か月後に酢になる。工業的な酢の製造過程では、酸素を供給することによってバクテリアによる酸化を促進する。
発酵によって酢が初めて作られたのは、おそらくワインの製造の失敗によるものである。 発酵中のブドウ果汁(ムスト)の熟成時に温度が高すぎると、アセトバクター属が自然にブドウに付着している酵母を圧倒してしまう。料理、医療、保健衛生における酢の需要が増すと、ワイン製造者たちはすぐに、ブドウが熟してワインの製造に適するようになる前の暑い夏季に他の有機物を使って酢を作ることを学んだ。しかし、ワイン製造者たちは発酵の過程を理解していなかったため、その方法は時間がかかる上にいつも成功するとは限らなかった[99]。
最初の近代的な工業的生産過程の1つは「促成法」あるいは「ジャーマン法」と呼ばれるもので、1823年にドイツで使われ始めた。この方法では、発酵は木の削り屑や炭を詰めた塔の中で行われる。エタノールを含んだ原料が塔の頂上から流し込まれ、新鮮な空気を自然に、または人為的な対流によって供給する。空気の供給量を増やすことによって、数ヶ月かかった酢の製造は数週間に短縮された[100]。
今日における酢の製造には1949年にオットー・ホロマツカとハインリヒ・エプナーによって考案された[101]浸水形の培養槽が用いられている。この方法では、発酵は撹拌されるタンクの中で溶液に酸素を通じさせながら行われ、15%の酢酸を含んだ酢が24時間で、流加培養法を使うと20%の濃度のものが60時間ででき上がる[99]。
無気性発酵
クロストリジウム属 (Clostridium) のある種の嫌気性バクテリアは糖類を直接酢酸に変換させることができ、中間体としてエタノールを必要としない。これらのバクテリアによる化学反応は全体として次のようなものである
C6H12O6⟶3CH3COOH{displaystyle {ce {C6H12O6 -> 3 CH3COOH}}}[102]
これらの酢酸産生菌の多くはメタノール、一酸化炭素、または二酸化炭素と水素の混合物など、1炭素の化合物から直接酢酸を作り出すことができる[103]。
- 2CO2+4H2⟶CH3COOH+2H2O{displaystyle {ce {2 CO2 + 4 H2 -> CH3COOH + 2 H2O}}}

糖類またはより安価な原料を直接酢酸の製造に利用できるクロストリジウム属の能力は、アセトバクター属のようなエタノール酸化菌より効率的に酢酸を作り出せる可能性があることを示している。しかしながら、クロストリジウム属は酸に弱く、最も酸に強いクロストリジウム属でも数%の酢酸を含む酢しか作れない。一方、アセトバクター属には酢酸濃度20%までの酢を作ることができるものがある。アセトバクター属を使う酢の製造はクロストリジウム属で作った酢を濃縮するよりも価格面でより効率的である。その結果、酢酸産生菌は1940年からその存在が知られているものの、工業的な利用はニッチな用途に限られている[104]。
用途

研究室で使われる 2.5 L 瓶入りの酢酸
多くの化合物を作る際に試薬として用いられる。主に酢酸ビニルモノマーの製造に使われ、無水酢酸や他の酢酸エステルがこれに次ぐ。酢として利用される酢酸は比較的少ない。
酢酸ビニルモノマー
酢酸の主要な用途は酢酸ビニルモノマーの製造である[84]。2003年、全世界で消費される酢酸のうち43.5%がこの目的で消費された[105]。酸素の存在下、エチレンと酢酸をパラジウム触媒で反応させることで得られる[106]。
- 2CH3COOH+2CH2=CH2+O2⟶2CH3COO−CH=CH2+2H2O{displaystyle {ce {2 CH3COOH + 2 CH2=CH2 + O2 -> 2CH3COO-CH=CH2 + 2 H2O}}}

酢酸ビニルは重合させてポリ酢酸ビニルなどのポリマーとしたのち、塗料や接着剤として使われる[107]。
無水酢酸
2分子の酢酸を脱水縮合させると無水酢酸が得られる。これは酢酸ビニルモノマー用途に次ぐ酢酸の主要な用途であり、2009年には世界の全消費量のうち18 %が無水酢酸の製造に使用されている[84]。酢酸メチルのカルボニル化によって直接得ることもでき[108]、カティバ法のプラントをこの目的に使うこともできる。
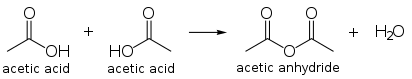
無水酢酸は強力なアセチル化試剤であり[109]、写真フィルムや合成繊維などの用途があるアセチルセルロースの製造などに用いられる[110]。またアスピリン[111]やヘロイン[112]などの合成にも使われる。
エステルの製造
酢酸のエステル類はインク、塗料、上塗の溶媒として使用される。酢酸エチル、酢酸ブチル、酢酸イソブチル、酢酸プロピルが一般的で、これらは対応するアルコールとの触媒反応によって合成される。
CH3COOH+HOR⟷CH3COO−R+H2O{displaystyle {ce {CH3COOH + HOR <-> CH3COO-R + H2O}}}[113](R はアルキル基を示す)
しかしながら、酢酸エステルの製造法としてはアルデヒドを原料としたティシチェンコ反応による合成が主流となっており、これは原料となるアルデヒドがアルコールよりも安価なためである[113]。また、エーテル類の酢酸エステルはニトロセルロース、アクリル塗料、ワニスの洗浄剤、木材用塗料の溶媒として使われる。まずグリコールのモノエーテルをエチレンオキシドやプロピレンオキシドとアルコールの反応で作り、これを酢酸でエステル化する。主なものはエチレングリコールモノエチルエーテル酢酸エステル (EEA)、エチレングリコールモノブチルエーテル酢酸エステル (EBA)、プロピレングリコールモノメチルエーテル酢酸エステル (PMA) の3つである。この用途には全生産量の17 %が消費される[84]。これらのエステルのうちいくつかは動物実験において生殖・発生毒性が示されており、例えばEEAではラットに対してEEAを経口投与した試験において受胎率の低下やオスの精子数の減少といった生殖毒性や、胎児の骨格奇形のような発生毒性などが確認されている[114]。そのため、EEAはリスクフレーズにおいてR60/R61(生殖毒性、胎児毒性)が指定されている[115]。
酢
酢には通常4-8 %、最大18 %の濃度の酢酸が含まれており、調味料や防腐剤として古くから利用されてきた[116]。2010年の日本の食酢の生産量は醸造酢が約41万キロリットル、合成酢が1400キロリットルであった[117]。また韓国では、氷酢酸がのり巻きや刺し身のたれを作る材料として食用に販売されている[118]。
溶媒
氷酢酸は優れた極性プロトン性溶媒であり、有機化合物の再結晶溶媒としてしばしば使われる。純粋な酢酸は、ポリエチレンテレフタラート (PET) の原料であるテレフタル酸の製造の際に溶媒として用いられる。2009年のPET製造用途における酢酸の消費量は世界の全消費量のうちの17 %を占めており、無水酢酸製造や酢酸エステル製造用途における消費量と同程度である[84]。
フリーデル・クラフツ反応などのようにカルボカチオンを含む反応にしばしば用いられる。例えば、樟脳の工業的製造の1工程はカンフェンのワーグナー・メーヤワイン転位による酢酸イソボルニルの生成だが、酢酸はこの際に転位生成物であるカルボカチオンのトラップ剤兼溶媒として働く[119]。パラジウム炭素を用いたベンジル基の脱保護においても、反応を促進させるための酸性溶媒として酢酸が選択される[120]。
分析化学においては、アニリンなどの弱い塩基の定量の際に用いられる。通常、アニリンのような弱塩基は水溶液中での解離度が低いため強酸による中和滴定を行うことができないが、水よりもプロトン供与能の高い酢酸中であれば強い塩基としてふるまい完全に解離することができる。一方で、過塩素酸は酢酸溶媒中においても強酸としてふるまうことができるため、酢酸溶媒中で弱塩基を過塩素酸で滴定することができる。このような酢酸を溶媒として用いた中和滴定は日本薬局方において多くの弱塩基性医薬品の定量方法として利用されている[121]。
その他
酢酸は代表的な弱酸である[122]。写真の現像において現像処理と定着処理の間で使われるが、これは現像液がアルカリ性であるから、弱い酸性を示す酢酸で現像処理を停止させるためである[123]。他に、カルシウムやマグネシウムなどによる水垢を除去するための洗浄剤[124]、クラゲに刺された場合すぐに塗布する事によって刺胞を不活性化し症状を和らげる治療薬[125]、軽度の外耳炎の治療[126]、といった用途があげられる。また、家畜用の牧草を保管するために作られるサイレージにおいては、牧草が酢酸発酵することで生成される酢酸によってpHが4程度まで低下することでバクテリアやカビの増殖が抑えられる[127]。コルポスコピー・上部消化管内視鏡においては粘膜を刺激し、正常粘膜と異常粘膜の反応の差異を判断に用いることがある[128]。氷酢酸、ピクリン酸、ホルマリンの混合溶液はブアン固定液として細胞の固定に利用される[129]。
様々な無機塩・有機塩類が酢酸から合成される。
酢酸カリウム - 融雪剤[130]。
酢酸ナトリウム - 媒染剤[131]、食品の日持向上剤[132]、エコカイロ(三水和物)[131]
酢酸銅(II) - 顔料[133]、殺菌剤[133]
酢酸アルミニウム、酢酸鉄(II) - 染料の媒染剤[134]
酢酸パラジウム(II) - ヘック反応など、カップリング反応の触媒[135]
酢酸の誘導体には以下のようなものがある。
モノクロロ酢酸 (MCA)、ジクロロ酢酸、トリクロロ酢酸 - MCA はインディゴ染料の製造に使われる[136]
ブロモ酢酸 - 医薬品や農薬などの合成原料[137]
トリフルオロ酢酸 - 有機合成における一般的な試薬[138]
出典
^ Cheung, Hosea; Tanke, Robin S.; Torrence, G. Paul (2005). “Acetic Acid”. Ullmann's Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. doi:10.1002/14356007.a01_045.
- ^ abcdefPayen, Anselme M. (1878). Paul, B. H.. ed. Industrial chemistry: A manual for use in technical colleges or schools and for manufacturers, etc. London: Longmans, Green, and Co. pp. 961–962. http://books.google.co.jp/books?id=GR9DAAAAIAAJ&pg=PA961.
^ Church, Arthur Herbert (1890). The chemistry of paints and painting. London: Seeley. p. 111. http://books.google.co.jp/books?id=a4c6AAAAMAAJ&pg=PA111.
^ Great Britain Patent Office (1871). Patents for inventions: abridgments of specifications. London: Office of the Commissioners of Patents for Inventons. p. p. xx.
- ^ abThomson, Robert Dundas (1854). Cyclopaedia of chemistry with its applications to mineralogy, physiology. London and Glasgow: Griffin and Co.. p. 3. http://books.google.co.jp/books?id=8tDnAAAAMAAJ&pg=PA3.
^ 村田徳治 『廃棄物のやさしい化学』 第1巻、日報出版、2009年、52頁。ISBN 4-89086-180-7。
^ Rezende, Lisa (2006). Chronology of science. New York: Facts on File. p. 61. ISBN 0-8160-5342-1.
- ^ abOthmer, Donald F.; Kirk, Raymond E.; Kroschwitz, Jacqueline I.; Howe-Grant, Mary (1991). Kirk-Othmer encyclopedia of chemical technology. 1. New Jersey: Wiley. p. 121. ISBN 047152669X.
^ Griffiths, Ralph (1799年). “Memoir on the Acetic Acid. By P A. Adet”. The Monthly review 28: 558.
^ Adet, P. A. (1798年). “Mémoire sur l'acide acétique”. Annales de Chemie 27: 299–319.
^ Darracq, M. (1802年). “Obsercations on the Acetous and Acetic Acids”. Journals of the Royal Institution of Great Britain 1: 132.
^ Rocke, Alan J. (1993). The quiet revolution: Hermann Kolbe and the science of organic chemistry. Berkeley: University of California Press. pp. 59–60.
^ Goldwhite, Harold (2003年9月). “Short summary of the career of the German organic chemist, Hermann Kolbe”. The New Haven Section of the American Chemical Society 20 (3).
^ 山口、山本、田村 (2010)、2頁。
- ^ ab桜井 (2001)、2頁。
- ^ abcdeMartin, Geoffrey (1917). Industrial and Manufacturing Chemistry (Part 1, Organic ed.). London: Crosby Lockwood. pp. 330–31. http://babel.hathitrust.org/cgi/pt?view=image;size=100;id=mdp.39076005015040;page=root;seq=366;num=330.
^ McClure, David Courtney. “Kilkerran Pyroligneous Acid Works 1845 to 1945”. Ayrshire History. 2011年8月19日閲覧。
^ Almqvist, Ebbe (2003). History of industrial gases. Berlin: Springer. p. 118. ISBN 0306472775. ; Brown, William H.; Foote, Christopher S.; Iverson, Brent L.; Anslyn, Eric V. (2011). Organic Chemistry (6th edition ed.). Belmont. p. 660. ISBN 084005498X.
^ Aftalion, Fred (1991). A history of the international chemical industry. Philadelphia: University of Pennsylvania Press. p. 135.
^ 桜井 (2001)、3頁。
^ 紫垣明典 「酢酸製造プロセスの進歩」『有機化学合成協会誌』 第41巻、1983年、553–560頁。
^ 藤森萬年 『産業倫理』 文芸社、2006年、43頁。
^ 桜井 (2001)、8頁。
^ 桜井 (2001)、6頁。
^ 大森晋爾 (2000年12月). “舎密開教”. 会報 第43号. 岡山日独協会. 2011年8月19日閲覧。
^ マクマリー (2009)、740頁。
^ IUPAC (2004), “Chapter P-1 Nomenclature of Organic Compounds”, Preferred IUPAC Names, p. 4, http://old.iupac.org/reports/provisional/abstract04/BB-prs310305/Chapter1.pdf .
^ 貞廣知行 (2008年2月24日). “エタン誘導体”. 化合物命名法談義. 2011年8月19日閲覧。
- ^ abcdeStoddard, John Tappan (1918). Introduction to Organic Chemistry (2nd ed. ed.). Philadelphia: P. Blakiston's Son & Co.. p. 96. http://books.google.com/books?id=ZKW4-3S8ryIC&pg=PA96. Forgotten Books から2010年に再版されている。
^ 山口、山本、田村 (2010)、30頁。
- ^ abcdSenning (2007), p. 3.
^ Senning (2007), 2.
^ Hilmen, Eva-Katrine (2000年11月). “Separation of Azeotropic Mixtures: Tools for Analysis and Studies on Batch Distillation Operation”. Norwegian University of Science and Technology, dept. of Chemical Engineering. p. 13. 2011年8月20日閲覧。
^ Armarego, Wilfred L. F.; Chai, Christina L. L. (2009). Purification of Laboratory Chemicals (6th ed. ed.). Burlington: Elsevier. p. 40.
- ^ ab“Product Description. Acetic Acid, Glacial”. Celanese. 2011年8月20日閲覧。
^ Zieborak, K.; Olszewski, K. (1958). Bulletin de l'Academie Polonaise des Sciences. Serie des Sciences Chimiques Geologiques et Geographiques 6 (2): 3315–3322
^ Emsley, John (1986年2月13日). “The solution is the problem”. New Scientist: 33–37. http://books.google.co.jp/books?id=Gu6wFrdExO4C&pg=PA33.
- ^ abThorpe, Gary (2007). AP Chemistry (4th ed. ed.). Hoboken: John Wiley and Sons. p. 63. http://books.google.co.jp/books?id=peKfop1XI8AC&pg=PA63.
- ^ abマクマリー (2009)、742頁。
^ Karle, J.; Brockway, L. O. (1944年). “An Electron Diffraction Investigation of the Monomers and Dimers of Formic, Acetic and Trifluoroacetic Acids and the Dimer of Deuterium Acetate”. Journal of the American Chemical Society 66 (4): 574–584. doi:10.1021/ja01232a022.
^ Jones, R. E.; Templeton, D. H. (1958年). “The crystal structure of acetic acid”. Acta Crystallographica 11 (7): 484–487. doi:10.1107/S0365110X58001341.
^ Briggs, James M.; Nguyen, Toan B.; Jorgensen, William L. (1991年). “Monte Carlo simulations of liquid acetic acid and methyl acetate with the OPLS potential functions”. The Journal of Physcal Chemistry 95: 3315–22. doi:10.1021/j100161a065.
^ Davies, M. M.; Sutherland, G. B. B. M. (1938年). “The Infra‐Red Absorption of Carboxylic Acids in Solution I. Qualitative Features”. The Journal of Chemical Physics 6: 755–767. doi:10.1063/1.1750166.
^ Walker, James (1899). Introduction to Physical Chemistry. New York: Macmillan and Co.. p. 197. http://books.google.com/books?id=evU_p902bKYC&pg=PA197.
^ “Hydrgen Bonding”. Competition Science Vision 1 (8): 1164–1172. (1998年). http://books.google.com/books?id=F-gDAAAAMBAJ&pg=PA1164.
^ “Product Description. Methyl Acetate”. Celanese. 2011年8月20日閲覧。
^ Sorrell, Thomas N. (2006). Organic Chemistry (2nd ed. ed.). Sausalito: University Science Books. p. 69. http://books.google.com/books?id=txmp1aoCJp8C&pg=PA69.
- ^ ab山口、山本、田村 (2010)、32頁。
- ^ abマクマリー (2009)、743頁。
^ 地球環境化学夏休み宿題解答-1, 東京工業大学附属科学技術高等学校, http://www.hst.titech.ac.jp/~meb/answer05.pdf
^ 『Dissociation Constants of Organic Acids and Bases』 p.1
^ Schorlemmer, Carl (1874). A manual of the chemistry of the carbon compounds. London: Macmillan. p. 137. http://books.google.co.jp/books?id=Kjs3AAAAMAAJ&pg=PA137.
^ Craig, Bruce D.; Anderson, David S. (1995). Handbook of Corrosion Data (2nd ed. ed.). Russell Township, Ohio: ASM International. pp. pp. 88–89. http://books.google.co.jp/books?id=KXwgAZJBWb0C&lpg=PA11&pg=RA1-PT41#v=onepage&q&f=false.
^ Jain, Rahul (2005). Xam Idea - Science (6th ed. ed.). New Delhi: V. K. (India) Enterprises. p. 31. http://books.google.co.jp/books?id=nRLQcNzSyVAC&pg=PP41.
^ Pavia, Donald L. (2005). Introduction to Organic Laboratory Techniques. Belmont: Thomson Learning. pp. pp. 93–96. http://books.google.co.jp/books?id=ega5c11VHvkC&pg=PA93#.
^ Kozhevnikov, Ivan V. (2002). Catalysis by Polyoxometalates. Catalysts for Fine Chemical Synthesis. 2. Chichester: John Wiley & Sons. p. 178. http://books.google.co.jp/books?id=jaTXrZyGSuAC&pg=PA178.
- ^ abColeman, G. H.; Alvarado, A. M. (1923年). “Acetamide”. Organic Syntheses 3: 3. http://www.orgsyn.org/demo.aspx?prep=cv1p0003. ; Collective Volume, 1, pp. 3
- ^ abAhluwalia, V. K.; Aggarwal, R. (2000). Comprehensive Practical Organic Chemistry. Hyderabad: Universities Press (India). pp. pp. 14–15. http://books.google.co.jp/books?id=mnsKyupepQEC&pg=PA14.
^ マクマリー (2009)、781頁。
^ Chauvel, A.; Lefebvre, G. (1989). Petrochemical Processes. Vol. 2. Paris: Editions Technip. p. 59. http://books.google.co.jp/books?id=3fkEv6TDqpkC&pg=PA59.
^ Wittcoff, Harold; Reuben, B. G.; Plotkin, Jeffrey S. (2004). Industrial Organic Chemicals (2nd ed. ed.). Hoboken: John Wiley & Sons. pp. pp. 365–369. http://books.google.co.jp/books?id=4KHzc-nYPNsC&pg=PA365.
^ Kane, Robert; Draper, John William (1851). Elements of Chemistry. New York: Harper and Brothers. p. 564. http://books.google.co.jp/books?id=1KWzya73ITsC&pg=PA564.
^ Maki-Arvela, Paivi; Salmi, Tapio; Paatero, Erkki (1994年). “Kinetics of the Chlorination of Acetic Acid with Chlorine in the Presence of Chlorosulfonic Acid and Thionyl Chloride”. Industrial & Engineering Chemistry Research 33 (9): 2073–2083. doi:10.1021/ie00033a008.
^ Dalton, David R. (2011). Foundations of Organic Chemistry. Hoboken: John Wiley & Sons. pp. pp. 648–649. http://books.google.co.jp/books?id=2rxFRgp57_0C&pg=PA648.
- ^ ab泉屋信夫、野田耕作、下東康幸 『生物化学序説』 第2版、化学同人、1998年、129–135頁。
^ パウラ・Y・ブルース 『有機化学概説』 第2版、大船泰史、香月勗、西郷和彦、富岡清 監訳、化学同人、2010年、559頁。
^ マクマリー (2009)、1125–1135頁。
^ ブルース (2009)、1273–1275頁。
^ ブルース (2009)、1281–1282頁。
^ “EC 6.2.1.1”. IUBMB Enzyme Nomenclature. 2011年9月4日閲覧。
^ “EC 6.2.1.1”. IUBMB Enzyme Nomenclature. 2011年9月4日閲覧。
^ K・P・C・ボルハルト、N・E・ショアー 『現代有機化学』 第4版 下巻、古賀憲司、野依良治、村橋俊一 監訳、大嶌幸一郎、小田嶋和徳、小松満男、戸部義人 訳、化学同人、2004年、921–925頁。
^ マクマリー (2009)、1054–1073頁。
^ 畑山巧 『ベーシック生化学』 化学同人、2009年、149–151頁。
^ 米谷民雄 『食品中の化学物質と安全性』 日本食品衛生協会、2009年、28頁。
^ 湯川英明 監修 『CO2固定化・削減と有効利用』 CMC出版、CMCテクニカルライブラリー 地球環境シリーズ 第336巻、2009年、223–224頁。
^ 工藤俊章、大熊盛也 監修 『難培養微生物の利用技術』CMC出版、CMCテクニカルライブラリー バイオテクノロジーシリーズ 第344巻、2010年、107頁。
^ ジョージ・C・マクガヴァン 『完璧版昆虫の写真図鑑』 日本語版 野村周平 監修、日本ヴォーグ、2000年、219頁。
^ Myers, Richard L. (2007). The 100 most important chemical compounds. Westport: Greenwood. p. 3. http://books.google.co.jp/books?id=MwpQWcIKMzAC&pg=PA3.
^ Yoneda, Noriyki; Kusano, Satoru; Yasui, Makoto; Pujado, Peter; Wilcher, Steve (2001年). “Recent advances in processes and catalysts for the production of acetic acid”. Applied Catalysis A: General 221: 253–265. doi:10.1016/S0926-860X(01)00800-6.
^ マクマリー (2009)、739頁。
^ 経済産業省生産動態統計・生産・出荷・在庫統計平成20年年計による
^ "Production report". Chem. Eng. News (July 11, 2005), 67–76.
- ^ abcdeSuresh, Bala (2003). "Acetic Acid". CEH Report 602.5000, SRI International.
- ^ abcアルペ (2004), 188頁。
^ Wagner, F. S. (1978). "Acetic acid". In Kirk-Othmer Encyclopedia of Chemical Technology; Grayson, M., Ed.; New York: John Wiley & Sons.; 3rd edition.
- ^ abvan Santen, Rutger A.; van Leeuwen, Piet W. N. M.; Moulijin, Jacob A.; Averill, Bruce A. (1999). Catalysis: An Integrated Approach (2nd ed.). Amsterdam: Elsevier. p. 15. ISBN 0444829636.
^ アルペ (2004), 189–190頁。
^ アルペ (2004), 190–191頁。
^ Lancaster, Mike (2002). Green Chemistry: an Introductory Text. Cambridge: Royal Society of Chemistry. pp. pp. 262–266. ISBN 0-85404-620-8.
- ^ ab桜井 (2001)、3–4頁。
^ 桜井 (2001)、5頁。
^ アルペ (2004), 184頁。
^ アルペ (2004), 185頁。
^ アルペ (2004), 185–186頁。
^ Sano, Ken-ichi; Uchida, Hiroshi; Wakabayashi, Syoichirou (1999年). “A new process for acetic acid production by direct oxidation of ethylene”. Catalyst Surveys from Japan 3: 55–60. doi:10.1023/A:1019003230537.
^ 桜井 (2001)、7–8頁。
^ “休止状態の酢酸プラント廃棄も 昭和電工”, 大分合同新聞, (2009-08-01), http://www.oita-press.co.jp/localNews/2009_124908787945.html 2011年9月11日閲覧。
- ^ abHromatka, Otto; Ebner, Heinrich (1959年). “Vinegar by Submerged Oxidative Fermentation”. Industrial & Engineering Chemistry 51 (10): 1279–1280. doi:10.1021/ie50598a033.
^ Partridge, Everett P. (1931年). “Acetic Acid and Cellulose Acetate in the United States. A General Survey of Economic and Technical Developments”. Industrial & Engineering Chemistry 23 (5): 482–498. doi:10.1021/ie50257a005.
^ Hromatka, Otto; Ebner, Heinrich (1949年). “Investigations on vinegar fermentation: Generator for vinegar fermentation and aeration procedures”. Enzymologia 13: 369.
^ JP 2010239913, 坂志朗, "有機酸発酵および直接水素化分解によるアルコール類の製造方法"
^ アメリカ合衆国特許第5,173,429号
^ Jia Huey Sim, Azlina Harun Kamaruddin, Wei Sing Long and Ghasem Najafpour (2007年). “Clostridium aceticum—A potential organism in catalyzing carbon monoxide to acetic acid: Application of response surface methodology”. Enzyme and Microbial Technology 40 (5): 1234–1243. doi:10.1016/j.enzmictec.2006.09.017.
^ “松原市年産20万トン酢酸プロジェクト”. 中国吉林省 政府ネット. 2012年2月21日閲覧。
^ 足立、岩倉、馬場 (2004)、107頁。
^ ブルース (2009)、1399頁。
^ 足立、岩倉、馬場 (2004)、120–121頁。
^ 中原勝儼 『化学大辞典』1、化学大辞典編集委員会(編)、共立、1981年10月、縮刷版第26版、107-108頁。
^ 祖父江、友田 『化学大辞典』1、化学大辞典編集委員会(編)、共立、1981年10月、縮刷版第26版、108-109頁。
^ ロバート J. ウーレット 『ウーレット 有機化学』 高橋知義、橋元親夫、堀内昭、須田憲男 訳、化学同人、2002年、380頁。ISBN 4759809147。
^ ブルース (2009)、1444頁。
- ^ abKlaus Weissermel,Hans-Jürgen Arpe (2008). Industrial Organic Chemistry. John Wiley & Sons. p. 187. ISBN 3527614591.
^ “CERI有害性評価書”. 化学物質評価研究機構. 2012年8月21日閲覧。
^ “PRTR・MSDS対象物質ハザードデータ”. 製品評価技術基盤機構. 2012年8月21日閲覧。
^ “VINEGAR (pdf)”. Global AgriSystem. p. 1-2. 2012年3月6日閲覧。
^ “食酢の生産動向”. 全国食酢協会中央会 全国食酢公正取引委員会. 2012年3月6日閲覧。
^ “韓国産「氷酢酸」飲まないで…急性胃炎発症も”. 読売新聞 (2009年11月20日). 2009年11月22日時点のオリジナルよりアーカイブ。2012年3月5日閲覧。
^ Charles Sell (2007). “Ingredients for the Modern Perfumery Industry”. In Charles Sell. The Chemistry of Fragrances: From Perfumer to Consumer. Royal Society of Chemistry. p. 80. ISBN 0854048243.
^ Roger Arthur Sheldon,Isabel Arends,Ulf Hanefeld (2007). Green Chemistry and Catalysis. Wiley-VCH. p. 99. ISBN 352730715X.
^ 桜井弘 『薬学のための分析化学』 化学同人、1999年、142-143頁。ISBN 4759808418。
^ 増田芳男、澤田清 『理系のための基礎化学』 化学同人、2006年、126頁。ISBN 4759810552。
^ “中央精機のホログラフィ (pdf)”. 中央精機. p. 43. 2013年5月26日閲覧。
^ 大矢勝 『よくわかる最新洗浄・洗剤の基本と仕組み』 秀和システム、2011年、90-91頁。ISBN 4798031828。
^ “心肺蘇生と救急心血管治療のためのガイドライン 2010 (pdf)”. アメリカ心臓協会. p. 28. 2013年5月24日閲覧。
^ “外耳道炎”. メルクマニュアル18版 日本語版. 2013年5月24日閲覧。
^ “熱帯地域サイレージ調製・利用の手引き (pdf)”. 畜産技術協会. pp. 3-4. 2013年5月28日閲覧。
^ 植木實、猪木千春 (2001年). “研修医のための必修知識 B.産婦人科検査法 5.コルコスコピー”. 日産婦誌 53 (5): 1. http://www.jsog.or.jp/PDF/53/5305-069.pdf.
^ 小出剛 『マウス実験の基礎知識』 オーム社、2009年、74-75頁。ISBN 4274502171。
^ 主成分は酢酸カリウム、グリセリン・水。
- ^ ab“安全データシート(SDS) (pdf)”. 昭和化学. p. 4. 2013年5月27日閲覧。
^ 林裕造 『食品安全ハンドブック』 食品安全ハンドブック編集委員会、丸善、2010年、306-308頁。ISBN 4621081829。
- ^ ab“製品安全データシート 酢酸銅(II)”. 中央労働災害防止協会 安全衛生情報センター. 2013年5月27日閲覧。
^ 小林孝行、山本周治 (2008年). “地域農産物由来天然色素による染色技術の開発”. 愛知県産業技術研究所研究報告 (愛知県産業技術研究所) 7: 137. http://www.aichi-inst.jp/mikawa/research/report/mikawa_2008_03.pdf 2013年5月27日閲覧。.
^ 窪田周平 『創薬支援研究の展望』 鳥澤保廣・監修、シーエムシー出版、2008年、138-139頁。ISBN 4882319985。
^ “モノクロロ酢酸”. 日本化学物質安全・情報センター. p. 2. 2013年5月27日閲覧。
^ “ブロモ酢酸”. 厚生労働省. 2013年5月27日閲覧。
^ “Trifluoroacetic acid (TFA)試薬 製品仕様書”. シグマアルドリッチ. 2013年5月27日閲覧。
参考文献
- 足立吟也、岩倉千秋、馬場章夫 『新しい工業化学―環境との調和をめざして』、2004年。ISBN 4759809554。
- クラウス・ヴァイセルメル、ハンス=ユルゲン・アルペ 『工業有機化学』 向山光昭 監訳、東京化学同人、2004年、第5版。ISBN 4-80790605-4。
- 桜井秀樹ほか 『化学と社会』 岩波書店〈岩波講座 現代科学への入門 18〉、2001年。ISBN 4-00-011048-9。
- パウラ・Y・ブルース 『ブルース有機化学』下、大船泰史、香月勗、西郷和彦、富岡清 監訳、化学同人、2009年、第5版。ISBN 4759811699。
ジョン・マクマリー 『有機化学』 伊東椒、児玉三明、荻野敏夫、深澤義正、通元夫 訳、東京化学同人、2009年、第7版。ISBN 9784807906987(上)、ISBN 9784807906994(中)、ISBN 9784807907007(下)。- 山口良平、山本行男、田村類 『ベーシック有機化学』 化学同人、2010年、第2版。ISBN 4759814396。
Senning, Alexander (2007). Elsevier's Dictionary of Chemoetymology. Amsterdam: Elsevier. ISBN 0-444-52239-5.
関連項目
- 酢酸カーミン溶液
- 酢酸オルセイン溶液
- 酢酸ダーリア溶液
- 無水酢酸
- 石炭化学
- 酢酸菌
| ||||||||||||||||||
| ||||||||||