HIV
HIV
Jump to navigation
Jump to search
Human immunodeficiency virus | |
|---|---|
 | |
| Scanning electron micrograph of HIV-1 (in green) budding from cultured lymphocyte. Multiple round bumps on cell surface represent sites of assembly and budding of virions. | |
Virus classification | |
| Group: | Group VI (ssRNA-RT) |
| Order: | Ortervirales |
| Family: | Retroviridae |
| Subfamily: | Orthoretrovirinae |
| Genus: | Lentivirus |
| Species | |
| |
The human immunodeficiency virus (HIV) is a lentivirus (a subgroup of retrovirus) that causes HIV infection and over time acquired immunodeficiency syndrome (AIDS).[1][2] AIDS is a condition in humans in which progressive failure of the immune system allows life-threatening opportunistic infections and cancers to thrive. Without treatment, average survival time after infection with HIV is estimated to be 9 to 11 years, depending on the HIV subtype.[3] In most cases, HIV is a sexually transmitted infection and occurs by contact with or transfer of blood, pre-ejaculate, semen, and vaginal fluids. Non-sexual transmission can occur from an infected mother to her infant during pregnancy, during childbirth by exposure to her blood or vaginal fluid, and through breast milk.[4][5][6][7] Within these bodily fluids, HIV is present as both free virus particles and virus within infected immune cells.
HIV infects vital cells in the human immune system, such as helper T cells (specifically CD4+ T cells), macrophages, and dendritic cells.[8] HIV infection leads to low levels of CD4+ T cells through a number of mechanisms, including pyroptosis of abortively infected T cells,[9]apoptosis of uninfected bystander cells,[10] direct viral killing of infected cells, and killing of infected CD4+ T cells by CD8+ cytotoxic lymphocytes that recognize infected cells.[11] When CD4+ T cell numbers decline below a critical level, cell-mediated immunity is lost, and the body becomes progressively more susceptible to opportunistic infections, leading to the development of AIDS.
Contents
1 Virology
1.1 Classification
1.2 Structure and genome
1.3 Tropism
1.4 Replication cycle
1.4.1 Entry to the cell
1.4.2 Replication and transcription
1.4.3 Recombination
1.4.4 Assembly and release
1.5 Spread within the body
1.6 Genetic variability
2 Diagnosis
3 Research
4 Treatment
5 History
5.1 Discovery
5.2 Origins
6 See also
7 References
8 Further reading
9 External links
Virology
Classification
| Species | Virulence | Infectivity | Prevalence | Inferred origin |
|---|---|---|---|---|
| HIV-1 | High | High | Global | Common chimpanzee |
| HIV-2 | Lower | Low | West Africa | Sooty mangabey |
HIV is a member of the genus Lentivirus,[12] part of the family Retroviridae.[13] Lentiviruses have many morphologies and biological properties in common. Many species are infected by lentiviruses, which are characteristically responsible for long-duration illnesses with a long incubation period.[14] Lentiviruses are transmitted as single-stranded, positive-sense, enveloped RNA viruses. Upon entry into the target cell, the viral RNA genome is converted (reverse transcribed) into double-stranded DNA by a virally encoded enzyme, reverse transcriptase, that is transported along with the viral genome in the virus particle. The resulting viral DNA is then imported into the cell nucleus and integrated into the cellular DNA by a virally encoded enzyme, integrase, and host co-factors.[15] Once integrated, the virus may become latent, allowing the virus and its host cell to avoid detection by the immune system, for an indiscriminate amount of time.[16] The HIV virus can remain dormant in the human body for up to ten years after primary infection; during this period the virus does not cause symptoms. Alternatively, the integrated viral DNA may be transcribed, producing new RNA genomes and viral proteins, using host cell resources, that are packaged and released from the cell as new virus particles that will begin the replication cycle anew.
Two types of HIV have been characterized: HIV-1 and HIV-2. HIV-1 is the virus that was initially discovered and termed both lymphadenopathy associated virus (LAV) and human T-lymphotropic virus 3 (HTLV-III). HIV-1 is more virulent and more infective than HIV-2,[17] and is the cause of the majority of HIV infections globally. The lower infectivity of HIV-2, compared to HIV-1, implies that fewer of those exposed to HIV-2 will be infected per exposure. Due to its relatively poor capacity for transmission, HIV-2 is largely confined to West Africa.[18]
Structure and genome

Diagram of the HIV virion
HIV is different in structure from other retroviruses. It is roughly spherical[19] with a diameter of about 120 nm, around 60 times smaller than a red blood cell.[20] It is composed of two copies of positive-sense single-stranded RNA that codes for the virus's nine genes enclosed by a conical capsid composed of 2,000 copies of the viral protein p24.[21] The single-stranded RNA is tightly bound to nucleocapsid proteins, p7, and enzymes needed for the development of the virion such as reverse transcriptase, proteases, ribonuclease and integrase. A matrix composed of the viral protein p17 surrounds the capsid ensuring the integrity of the virion particle.[21]
This is, in turn, surrounded by the viral envelope, that is composed of the lipid bilayer taken from the membrane of a human host cell when the newly formed virus particle buds from the cell. The viral envelope contains proteins from the host cell and relatively few copies of the HIV Envelope protein,[21] which consists of a cap made of three molecules known as glycoprotein (gp) 120, and a stem consisting of three gp41 molecules that anchor the structure into the viral envelope.[22][23] The Envelope protein, encoded by the HIV env gene, allows the virus to attach to target cells and fuse the viral envelope with the target cell's membrane releasing the viral contents into the cell and initiating the infectious cycle.[22]
As the sole viral protein on the surface of the virus, the Envelope protein is a major target for HIV vaccine efforts.[24] Over half of the mass of the trimeric envelope spike is N-linked glycans. The density is high as the glycans shield the underlying viral protein from neutralisation by antibodies. This is one of the most densely glycosylated molecules known and the density is sufficiently high to prevent the normal maturation process of glycans during biogenesis in the endoplasmic and Golgi apparatus.[25][26] The majority of the glycans are therefore stalled as immature 'high-mannose' glycans not normally present on human glycoproteins that are secreted or present on a cell surface.[27] The unusual processing and high density means that almost all broadly neutralising antibodies that have so far been identified (from a subset of patients that have been infected for many months to years) bind to or, are adapted to cope with, these envelope glycans.[28]
The molecular structure of the viral spike has now been determined by X-ray crystallography[29] and cryogenic electron microscopy.[30] These advances in structural biology were made possible due to the development of stable recombinant forms of the viral spike by the introduction of an intersubunit disulphide bond and an isoleucine to proline mutation (radical replacement of an amino acid) in gp41.[31] The so-called SOSIP trimers not only reproduce the antigenic properties of the native viral spike, but also display the same degree of immature glycans as presented on the native virus.[32] Recombinant trimeric viral spikes are promising vaccine candidates as they display less non-neutralising epitopes than recombinant monomeric gp120, which act to suppress the immune response to target epitopes.[33]

Structure of the RNA genome of HIV-1
The RNA genome consists of at least seven structural landmarks (LTR, TAR, RRE, PE, SLIP, CRS, and INS), and nine genes (gag, pol, and env, tat, rev, nef, vif, vpr, vpu, and sometimes a tenth tev, which is a fusion of tat, env and rev), encoding 19 proteins. Three of these genes, gag, pol, and env, contain information needed to make the structural proteins for new virus particles.[21] For example, env codes for a protein called gp160 that is cut in two by a cellular protease to form gp120 and gp41. The six remaining genes, tat, rev, nef, vif, vpr, and vpu (or vpx in the case of HIV-2), are regulatory genes for proteins that control the ability of HIV to infect cells, produce new copies of virus (replicate), or cause disease.[21]
The two tat proteins (p16 and p14) are transcriptional transactivators for the LTR promoter acting by binding the TAR RNA element. The TAR may also be processed into microRNAs that regulate the apoptosis genes ERCC1 and IER3.[34][35] The rev protein (p19) is involved in shuttling RNAs from the nucleus and the cytoplasm by binding to the RRE RNA element. The vif protein (p23) prevents the action of APOBEC3G (a cellular protein that deaminates cytidine to uridine in the single-stranded viral DNA and/or interferes with reverse transcription[36]). The vpr protein (p14) arrests cell division at G2/M. The nef protein (p27) down-regulates CD4 (the major viral receptor), as well as the MHC class I and class II molecules.[37][38][39]
Nef also interacts with SH3 domains. The vpu protein (p16) influences the release of new virus particles from infected cells.[21] The ends of each strand of HIV RNA contain an RNA sequence called a long terminal repeat (LTR). Regions in the LTR act as switches to control production of new viruses and can be triggered by proteins from either HIV or the host cell. The Psi element is involved in viral genome packaging and recognized by gag and rev proteins. The SLIP element (TTTTTT) is involved in the frameshift in the gag-pol reading frame required to make functional pol.[21]
Tropism

Diagram of the immature and mature forms of HIV
The term viral tropism refers to the cell types a virus infects. HIV can infect a variety of immune cells such as CD4+ T cells, macrophages, and microglial cells. HIV-1 entry to macrophages and CD4+ T cells is mediated through interaction of the virion envelope glycoproteins (gp120) with the CD4 molecule on the target cells' membrane and also with chemokine co-receptors.[22][40]
Macrophage-tropic (M-tropic) strains of HIV-1, or non-syncytia-inducing strains (NSI; now called R5 viruses[41]) use the β-chemokine receptor, CCR5, for entry and are thus able to replicate in both macrophages and CD4+ T cells.[42] This CCR5 co-receptor is used by almost all primary HIV-1 isolates regardless of viral genetic subtype. Indeed, macrophages play a key role in several critical aspects of HIV infection. They appear to be the first cells infected by HIV and perhaps the source of HIV production when CD4+ cells become depleted in the patient. Macrophages and microglial cells are the cells infected by HIV in the central nervous system. In the tonsils and adenoids of HIV-infected patients, macrophages fuse into multinucleated giant cells that produce huge amounts of virus.
T-tropic strains of HIV-1, or syncytia-inducing strains (SI; now called X4 viruses[41]) replicate in primary CD4+ T cells as well as in macrophages and use the α-chemokine receptor, CXCR4, for entry.[42][43][44]
Dual-tropic HIV-1 strains are thought to be transitional strains of HIV-1 and thus are able to use both CCR5 and CXCR4 as co-receptors for viral entry.
The α-chemokine SDF-1, a ligand for CXCR4, suppresses replication of T-tropic HIV-1 isolates. It does this by down-regulating the expression of CXCR4 on the surface of HIV target cells. M-tropic HIV-1 isolates that use only the CCR5 receptor are termed R5; those that use only CXCR4 are termed X4, and those that use both, X4R5. However, the use of co-receptors alone does not explain viral tropism, as not all R5 viruses are able to use CCR5 on macrophages for a productive infection[42] and HIV can also infect a subtype of myeloid dendritic cells,[45] which probably constitute a reservoir that maintains infection when CD4+ T cell numbers have declined to extremely low levels.
Some people are resistant to certain strains of HIV.[46] For example, people with the CCR5-Δ32 mutation are resistant to infection by the R5 virus, as the mutation leaves HIV unable to bind to this co-receptor, reducing its ability to infect target cells.
Sexual intercourse is the major mode of HIV transmission. Both X4 and R5 HIV are present in the seminal fluid, which enables the virus to be transmitted from a male to his sexual partner. The virions can then infect numerous cellular targets and disseminate into the whole organism. However, a selection process[further explanation needed] leads to a predominant transmission of the R5 virus through this pathway.[47][48][49] In patients infected with subtype B HIV-1, there is often a co-receptor switch in late-stage disease and T-tropic variants that can infect a variety of T cells through CXCR4.[50] These variants then replicate more aggressively with heightened virulence that causes rapid T cell depletion, immune system collapse, and opportunistic infections that mark the advent of AIDS.[51] Thus, during the course of infection, viral adaptation to the use of CXCR4 instead of CCR5 may be a key step in the progression to AIDS. A number of studies with subtype B-infected individuals have determined that between 40 and 50 percent of AIDS patients can harbour viruses of the SI and, it is presumed, the X4 phenotypes.[52][53]
HIV-2 is much less pathogenic than HIV-1 and is restricted in its worldwide distribution to West Africa. The adoption of "accessory genes" by HIV-2 and its more promiscuous pattern of co-receptor usage (including CD4-independence) may assist the virus in its adaptation to avoid innate restriction factors present in host cells. Adaptation to use normal cellular machinery to enable transmission and productive infection has also aided the establishment of HIV-2 replication in humans. A survival strategy for any infectious agent is not to kill its host, but ultimately become a commensal organism. Having achieved a low pathogenicity, over time, variants that are more successful at transmission will be selected.[54]
Replication cycle

The HIV replication cycle
Entry to the cell

Mechanism of viral entry: 1. Initial interaction between gp120 and CD4. 2. Conformational change in gp120 allows for secondary interaction with CCR5. 3. The distal tips of gp41 are inserted into the cellular membrane. 4. gp41 undergoes significant conformational change; folding in half and forming coiled-coils. This process pulls the viral and cellular membranes together, fusing them.
The HIV virion enters macrophages and CD4+T cells by the adsorption of glycoproteins on its surface to receptors on the target cell followed by fusion of the viral envelope with the target cell membrane and the release of the HIV capsid into the cell.[55][56]
Entry to the cell begins through interaction of the trimeric envelope complex (gp160 spike) on the HIV viral envelope and both CD4 and a chemokine co-receptor (generally either CCR5 or CXCR4, but others are known to interact) on the target cell surface.[55][56] Gp120 binds to integrin α4β7 activating LFA-1, the central integrin involved in the establishment of virological synapses, which facilitate efficient cell-to-cell spreading of HIV-1.[57] The gp160 spike contains binding domains for both CD4 and chemokine receptors.[55][56]
The first step in fusion involves the high-affinity attachment of the CD4 binding domains of gp120 to CD4. Once gp120 is bound with the CD4 protein, the envelope complex undergoes a structural change, exposing the chemokine receptor binding domains of gp120 and allowing them to interact with the target chemokine receptor.[55][56] This allows for a more stable two-pronged attachment, which allows the N-terminal fusion peptide gp41 to penetrate the cell membrane.[55][56]Repeat sequences in gp41, HR1, and HR2 then interact, causing the collapse of the extracellular portion of gp41 into a hairpin shape. This loop structure brings the virus and cell membranes close together, allowing fusion of the membranes and subsequent entry of the viral capsid.[55][56]
After HIV has bound to the target cell, the HIV RNA and various enzymes, including reverse transcriptase, integrase, ribonuclease, and protease, are injected into the cell.[55][not in citation given] During the microtubule-based transport to the nucleus, the viral single-strand RNA genome is transcribed into double-strand DNA, which is then integrated into a host chromosome.
HIV can infect dendritic cells (DCs) by this CD4-CCR5 route, but another route using mannose-specific C-type lectin receptors such as DC-SIGN can also be used.[58] DCs are one of the first cells encountered by the virus during sexual transmission. They are currently thought to play an important role by transmitting HIV to T cells when the virus is captured in the mucosa by DCs.[58] The presence of FEZ-1, which occurs naturally in neurons, is believed to prevent the infection of cells by HIV.[59]

Clathrin-mediated endocytosis
HIV-1 entry, as well as entry of many other retroviruses, has long been believed to occur exclusively at the plasma membrane. More recently, however, productive infection by pH-independent, clathrin-mediated endocytosis of HIV-1 has also been reported and was recently suggested to constitute the only route of productive entry.[60][61][62][63][64]
Replication and transcription
Reverse transcription of the HIV genome into double-stranded DNA
Shortly after the viral capsid enters the cell, an enzyme called reverse transcriptase liberates the positive-sense single-stranded RNA genome from the attached viral proteins and copies it into a complementary DNA (cDNA) molecule.[65] The process of reverse transcription is extremely error-prone, and the resulting mutations may cause drug resistance or allow the virus to evade the body's immune system. The reverse transcriptase also has ribonuclease activity that degrades the viral RNA during the synthesis of cDNA, as well as DNA-dependent DNA polymerase activity that creates a sense DNA from the antisense cDNA.[66] Together, the cDNA and its complement form a double-stranded viral DNA that is then transported into the cell nucleus. The integration of the viral DNA into the host cell's genome is carried out by another viral enzyme called integrase.[65]
The integrated viral DNA may then lie dormant, in the latent stage of HIV infection.[65] To actively produce the virus, certain cellular transcription factors need to be present, the most important of which is NF-κB (nuclear factor kappa B), which is upregulated when T cells become activated.[67] This means that those cells most likely to be targeted, entered and subsequently killed by HIV are those actively fighting infection.
During viral replication, the integrated DNA provirus is transcribed into RNA, some of which then undergo RNA splicing to produce mature messenger RNAs (mRNAs). These mRNAs are exported from the nucleus into the cytoplasm, where they are translated into the regulatory proteins Tat (which encourages new virus production) and Rev. As the newly produced Rev protein is produced it moves to the nucleus, where it binds to full-length, unspliced copies of virus RNAs and allows them to leave the nucleus.[68] Some of these full-length RNAs function as new copies of the virus genome, while others function as mRNAs that are translated to produce the structural proteins Gag and Env. Gag proteins bind to copies of the virus RNA genome to package them into new virus particles.[69]
HIV-1 and HIV-2 appear to package their RNA differently.[70][citation needed] HIV-1 will bind to any appropriate RNA.[citation needed] HIV-2 will preferentially bind to the mRNA that was used to create the Gag protein itself.[71]
Recombination
Two RNA genomes are encapsidated in each HIV-1 particle (see Structure and genome of HIV). Upon infection and replication catalyzed by reverse transcriptase, recombination between the two genomes can occur.[72][73] Recombination occurs as the single-strand, positive-sense RNA genomes are reverse transcribed to form DNA. During reverse transcription, the nascent DNA can switch multiple times between the two copies of the viral RNA. This form of recombination is known as copy-choice. Recombination events may occur throughout the genome. Anywhere from two to 20 recombination events per genome may occur at each replication cycle, and these events can rapidly shuffle the genetic information that is transmitted from parental to progeny genomes.[73]
Viral recombination produces genetic variation that likely contributes to the evolution of resistance to anti-retroviral therapy.[74] Recombination may also contribute, in principle, to overcoming the immune defenses of the host. Yet, for the adaptive advantages of genetic variation to be realized, the two viral genomes packaged in individual infecting virus particles need to have arisen from separate progenitor parental viruses of differing genetic constitution. It is unknown how often such mixed packaging occurs under natural conditions.[75]
Bonhoeffer et al.[76] suggested that template switching by reverse transcriptase acts as a repair process to deal with breaks in the single-stranded RNA genome. In addition, Hu and Temin[72] suggested that recombination is an adaptation for repair of damage in the RNA genomes. Strand switching (copy-choice recombination) by reverse transcriptase could generate an undamaged copy of genomic DNA from two damaged single-stranded RNA genome copies. This view of the adaptive benefit of recombination in HIV could explain why each HIV particle contains two complete genomes, rather than one. Furthermore, the view that recombination is a repair process implies that the benefit of repair can occur at each replication cycle, and that this benefit can be realized whether or not the two genomes differ genetically. On the view that recombination in HIV is a repair process, the generation of recombinational variation would be a consequence, but not the cause of, the evolution of template switching.[76]
HIV-1 infection causes chronic inflammation and production of reactive oxygen species.[77] Thus, the HIV genome may be vulnerable to oxidative damages, including breaks in the single-stranded RNA. For HIV, as well as for viruses in general, successful infection depends on overcoming host defensive strategies that often include production of genome-damaging reactive oxygen species. Thus, Michod et al.[78] suggested that recombination by viruses is an adaptation for repair of genome damages, and that recombinational variation is a byproduct that may provide a separate benefit.
Assembly and release

HIV assembling on the surface of an infected macrophage. The HIV virions have been marked with a green fluorescent tag and then viewed under a fluorescent microscope.
The final step of the viral cycle, assembly of new HIV-1 virions, begins at the plasma membrane of the host cell. The Env polyprotein (gp160) goes through the endoplasmic reticulum and is transported to the Golgi apparatus where it is cleaved by furin resulting in the two HIV envelope glycoproteins, gp41 and gp120.[79] These are transported to the plasma membrane of the host cell where gp41 anchors gp120 to the membrane of the infected cell. The Gag (p55) and Gag-Pol (p160) polyproteins also associate with the inner surface of the plasma membrane along with the HIV genomic RNA as the forming virion begins to bud from the host cell. The budded virion is still immature as the gag polyproteins still need to be cleaved into the actual matrix, capsid and nucleocapsid proteins. This cleavage is mediated by the packaged viral protease and can be inhibited by antiretroviral drugs of the protease inhibitor class. The various structural components then assemble to produce a mature HIV virion.[80] Only mature virions are then able to infect another cell.
Spread within the body

Animation demonstrating cell-free spread of HIV.
The classical process of infection of a cell by a virion can be called "cell-free spread" to distinguish it from a more recently recognized process called "cell-to-cell spread".[81] In cell-free spread (see figure), virus particles bud from an infected T cell, enter the blood or extracellular fluid and then infect another T cell following a chance encounter.[81] HIV can also disseminate by direct transmission from one cell to another by a process of cell-to-cell spread, for which two pathways have been described. Firstly, an infected T cell can transmit virus directly to a target T cell via a virological synapse.[57][82] Secondly, an antigen-presenting cell (APC), such as a macrophage or dendritic cell, can transmit HIV to T cells by a process that either involves productive infection (in the case of macrophages) or capture and transfer of virions in trans (in the case of dendritic cells).[83] Whichever pathway is used, infection by cell-to-cell transfer is reported to be much more efficient than cell-free virus spread.[84] A number of factors contribute to this increased efficiency, including polarised virus budding towards the site of cell-to-cell contact, close apposition of cells, which minimizes fluid-phase diffusion of virions, and clustering of HIV entry receptors on the target cell towards the contact zone.[82] Cell-to-cell spread is thought to be particularly important in lymphoid tissues where CD4+ T cells are densely packed and likely to interact frequently.[81] Intravital imaging studies have supported the concept of the HIV virological synapse in vivo.[85] The many spreading mechanisms available to HIV contribute to the virus' ongoing replication in spite of anti-retroviral therapies.[81][86]
Genetic variability

The phylogenetic tree of the SIV and HIV
HIV differs from many viruses in that it has very high genetic variability. This diversity is a result of its fast replication cycle, with the generation of about 1010 virions every day, coupled with a high mutation rate of approximately 3 x 10−5 per nucleotide base per cycle of replication and recombinogenic properties of reverse transcriptase.[87][88][89]
This complex scenario leads to the generation of many variants of HIV in a single infected patient in the course of one day.[87] This variability is compounded when a single cell is simultaneously infected by two or more different strains of HIV. When simultaneous infection occurs, the genome of progeny virions may be composed of RNA strands from two different strains. This hybrid virion then infects a new cell where it undergoes replication. As this happens, the reverse transcriptase, by jumping back and forth between the two different RNA templates, will generate a newly synthesized retroviral DNA sequence that is a recombinant between the two parental genomes.[87] This recombination is most obvious when it occurs between subtypes.[87]
The closely related simian immunodeficiency virus (SIV) has evolved into many strains, classified by the natural host species. SIV strains of the African green monkey (SIVagm) and sooty mangabey (SIVsmm) are thought to have a long evolutionary history with their hosts. These hosts have adapted to the presence of the virus,[90] which is present at high levels in the host's blood, but evokes only a mild immune response,[91] does not cause the development of simian AIDS,[92] and does not undergo the extensive mutation and recombination typical of HIV infection in humans.[93]
In contrast, when these strains infect species that have not adapted to SIV ("heterologous" or similar hosts such as rhesus or cynomologus macaques), the animals develop AIDS and the virus generates genetic diversity similar to what is seen in human HIV infection.[94]Chimpanzee SIV (SIVcpz), the closest genetic relative of HIV-1, is associated with increased mortality and AIDS-like symptoms in its natural host.[95] SIVcpz appears to have been transmitted relatively recently to chimpanzee and human populations, so their hosts have not yet adapted to the virus.[90] This virus has also lost a function of the nef gene that is present in most SIVs. For non-pathogenic SIV variants, nef suppresses T cell activation through the CD3 marker. Nef's function in non-pathogenic forms of SIV is to downregulate expression of inflammatory cytokines, MHC-1, and signals that affect T cell trafficking. In HIV-1 and SIVcpz, nef does not inhibit T-cell activation and it has lost this function. Without this function, T cell depletion is more likely, leading to immunodeficiency.[95][96]
Three groups of HIV-1 have been identified on the basis of differences in the envelope (env) region: M, N, and O.[97] Group M is the most prevalent and is subdivided into eight subtypes (or clades), based on the whole genome, which are geographically distinct.[98] The most prevalent are subtypes B (found mainly in North America and Europe), A and D (found mainly in Africa), and C (found mainly in Africa and Asia); these subtypes form branches in the phylogenetic tree representing the lineage of the M group of HIV-1. Co-infection with distinct subtypes gives rise to circulating recombinant forms (CRFs). In 2000, the last year in which an analysis of global subtype prevalence was made, 47.2% of infections worldwide were of subtype C, 26.7% were of subtype A/CRF02_AG, 12.3% were of subtype B, 5.3% were of subtype D, 3.2% were of CRF_AE, and the remaining 5.3% were composed of other subtypes and CRFs.[99] Most HIV-1 research is focused on subtype B; few laboratories focus on the other subtypes.[100] The existence of a fourth group, "P", has been hypothesised based on a virus isolated in 2009.[101] The strain is apparently derived from gorilla SIV (SIVgor), first isolated from western lowland gorillas in 2006.[101]
HIV-2's closest relative is SIVsm, a strain of SIV found in sooty mangabees. Since HIV-1 is derived from SIVcpz, and HIV-2 from SIVsm, the genetic sequence of HIV-2 is only partially homologous to HIV-1 and more closely resembles that of SIVsm.[citation needed][102]
Diagnosis
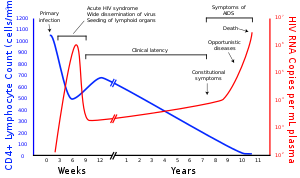
A generalized graph of the relationship between HIV copies (viral load) and CD4 counts over the average course of untreated HIV infection; any particular individual's disease course may vary considerably.
CD4+ T cell count (cells per µL)
HIV RNA copies per mL of plasma
Many HIV-positive people are unaware that they are infected with the virus.[103] For example, in 2001 less than 1% of the sexually active urban population in Africa had been tested, and this proportion is even lower in rural populations.[103] Furthermore, in 2001 only 0.5% of pregnant women attending urban health facilities were counselled, tested or receive their test results.[103] Again, this proportion is even lower in rural health facilities.[103] Since donors may therefore be unaware of their infection, donor blood and blood products used in medicine and medical research are routinely screened for HIV.[104]
HIV-1 testing is initially done using an enzyme-linked immunosorbent assay (ELISA) to detect antibodies to HIV-1. Specimens with a non-reactive result from the initial ELISA are considered HIV-negative, unless new exposure to an infected partner or partner of unknown HIV status has occurred. Specimens with a reactive ELISA result are retested in duplicate.[105] If the result of either duplicate test is reactive, the specimen is reported as repeatedly reactive and undergoes confirmatory testing with a more specific supplemental test (e.g., a polymerase chain reaction (PCR), western blot or, less commonly, an immunofluorescence assay (IFA)). Only specimens that are repeatedly reactive by ELISA and positive by IFA or PCR or reactive by western blot are considered HIV-positive and indicative of HIV infection. Specimens that are repeatedly ELISA-reactive occasionally provide an indeterminate western blot result, which may be either an incomplete antibody response to HIV in an infected person or nonspecific reactions in an uninfected person.[106]
HIV deaths (other than U.S.) in 2014.[107]
Nigeria (15.76%)
South Africa (12.51%)
India (11.50%)
Tanzania (4.169%)
Mozambique (4.061%)
Zimbabwe (3.49%)
Cameroon (3.09%)
Indonesia (3.04%)
Kenya (2.98%)
Uganda (2.97%)
Malawi (2.94%)
DR Congo (2.17%)
Ethiopia (2.11%)
Other (29.21%)
Although IFA can be used to confirm infection in these ambiguous cases, this assay is not widely used. In general, a second specimen should be collected more than a month later and retested for persons with indeterminate western blot results. Although much less commonly available, nucleic acid testing (e.g., viral RNA or proviral DNA amplification method) can also help diagnosis in certain situations.[105] In addition, a few tested specimens might provide inconclusive results because of a low quantity specimen. In these situations, a second specimen is collected and tested for HIV infection.
Modern HIV testing is extremely accurate, when the window period is taken into consideration. A single screening test is correct more than 99% of the time.[108] The chance of a false-positive result in a standard two-step testing protocol is estimated to be about 1 in 250,000 in a low risk population.[109] Testing post-exposure is recommended immediately and then at six weeks, three months, and six months.[110]
The latest recommendations of the US Centers for Disease Control and Prevention (CDC) show that HIV testing must start with an immunoassay combination test for HIV-1 and HIV-2 antibodies and p24 antigen. A negative result rules out HIV exposure, while a positive one must be followed by an HIV-1/2 antibody differentiation immunoassay to detect which antibodies are present. This gives rise to four possible scenarios:
- 1. HIV-1 (+) & HIV-2 (−): HIV-1 antibodies detected
- 2. HIV-1 (−) & HIV-2 (+): HIV-2 antibodies detected
- 3. HIV-1 (+) & HIV-2 (+): both HIV-1 and HIV-2 antibodies detected
- 4. HIV-1 (−) or indeterminate & HIV-2 (−): Nucleic acid test must be carried out to detect the acute infection of HIV-1 or its absence.[111]
Research
HIV/AIDS research includes all medical research that attempts to prevent, treat, or cure HIV/AIDS, as well as fundamental research about the nature of HIV as an infectious agent and AIDS as the disease caused by HIV.
Many governments and research institutions participate in HIV/AIDS research. This research includes behavioral health interventions, such as research into sex education, and drug development, such as research into microbicides for sexually transmitted diseases, HIV vaccines, and anti-retroviral drugs.[112] Other medical research areas include the topics of pre-exposure prophylaxis, post-exposure prophylaxis, circumcision and HIV, and accelerated aging effects.
After many years of research, an untested HIV vaccine has been created.[113] Bi-specific antibodies, that target both the surface of T-cells and viral epitopes, can prevent entry of the virus into human cells.[114] Another group has utilised the same technology to develop a bi-specific antibody that neutralises viral particles by cross-linking of envelope glycoproteins.[115]
Treatment
HIV latency, and the consequent viral reservoir in CD4+ T cells, dendritic cells, as well as macrophages, is the main barrier to eradication of the virus.[16]
It is important to note that although HIV is highly virulent, transmission is greatly reduced when an HIV-infected person has a suppressed or undetectable viral load (<50 copies/ml) due to prolonged and successful anti-retroviral treatment. Hence, it can be said to be almost impossible (but still non-zero) for an HIV-infected person who has an undetectable viral load to transmit the virus, even during unprotected sexual intercourse, as there would be a negligible amount of HIV present in the seminal fluid, vaginal secretions or blood, for transmission to occur.[116][117] This does not mean, however, that prolonged anti-retroviral treatment will result in a suppressed viral load. An undetectable viral load, generally agreed as less than 50 copies per milliliter of blood, can only be proven by a polymerase chain reaction (PCR) test.[118]
At the same time, it is important to recognise that reaching an undetectable viral load is determined by many factors, including treatment adherence, HIV resistance to certain anti-retroviral drugs, stigma, and inadequate health systems.[119]
History
Discovery
AIDS was first clinically observed in 1981 in the United States.[120] The initial cases were a cluster of injection drug users and gay men with no known cause of impaired immunity who showed symptoms of Pneumocystis jirovecii pneumonia (PJP), a rare opportunistic infection that was known to occur in people with very compromised immune systems.[121] Soon thereafter, additional gay men developed a previously rare skin cancer called Kaposi's sarcoma (KS).[122][123] Many more cases of PJP and KS emerged, alerting U.S. Centers for Disease Control and Prevention (CDC) and a CDC task force was formed to monitor the outbreak.[124] The earliest retrospectively described case of AIDS is believed to have been in Norway beginning in 1966.[125]
In the beginning, the CDC did not have an official name for the disease, often referring to it by way of the diseases that were associated with it, for example, lymphadenopathy, the disease after which the discoverers of HIV originally named the virus.[126][127] They also used Kaposi's Sarcoma and Opportunistic Infections, the name by which a task force had been set up in 1981.[128] In the general press, the term GRID, which stood for gay-related immune deficiency, had been coined.[129] The CDC, in search of a name, and looking at the infected communities coined "the 4H disease", as it seemed to single out homosexuals, heroin users, hemophiliacs, and Haitians.[130][131] However, after determining that AIDS was not isolated to the gay community,[128] it was realized that the term GRID was misleading and AIDS was introduced at a meeting in July 1982.[132] By September 1982 the CDC started using the name AIDS.[133]

Françoise Barré-Sinoussi, co-discoverer of HIV
In 1983, two separate research groups led by American Robert Gallo and French investigators Françoise Barré-Sinoussi and Luc Montagnier independently declared that a novel retrovirus may have been infecting AIDS patients, and published their findings in the same issue of the journal Science.[134][135][136] Gallo claimed that a virus his group had isolated from a person with AIDS was strikingly similar in shape to other human T-lymphotropic viruses (HTLVs) his group had been the first to isolate. Gallo's group called their newly isolated virus HTLV-III. At the same time, Montagnier's group isolated a virus from a patient presenting with swelling of the lymph nodes of the neck and physical weakness, two classic symptoms of primary HIV infection. Contradicting the report from Gallo's group, Montagnier and his colleagues showed that core proteins of this virus were immunologically different from those of HTLV-I. Montagnier's group named their isolated virus lymphadenopathy-associated virus (LAV).[124] As these two viruses turned out to be the same, in 1986 LAV and HTLV-III were renamed HIV.[137]
Another group working contemporaneously with the Montagnier and Gallo groups was that of Dr. Jay Levy at the University of California, San Francisco. He independently discovered the AIDS virus in 1983 and named it the AIDS associated retrovirus (ARV).[138] This virus was very different from the virus reported by the Montagnier and Gallo groups. The ARV strains indicated, for the first time, the heterogeneity of HIV isolates and several of these remain classic examples of the AIDS virus found in the United States.[139]
Origins
Both HIV-1 and HIV-2 are believed to have originated in non-human primates in West-central Africa, and are believed to have transferred to humans (a process known as zoonosis) in the early 20th century.[140][141]
HIV-1 appears to have originated in southern Cameroon through the evolution of SIVcpz, a simian immunodeficiency virus (SIV) that infects wild chimpanzees (HIV-1 descends from the SIVcpz endemic in the chimpanzee subspecies Pan troglodytes troglodytes).[142][143] The closest relative of HIV-2 is SIVsmm, a virus of the sooty mangabey (Cercocebus atys atys), an Old World monkey living in littoral West Africa (from southern Senegal to western Côte d'Ivoire).[18]New World monkeys such as the owl monkey are resistant to HIV-1 infection, possibly because of a genomic fusion of two viral resistance genes.[144]
HIV-1 is thought to have jumped the species barrier on at least three separate occasions, giving rise to the three groups of the virus, M, N, and O.[145]

Left to right: the African green monkey source of SIV, the sooty mangabey source of HIV-2, and the chimpanzee source of HIV-1
There is evidence that humans who participate in bushmeat activities, either as hunters or as bushmeat vendors, commonly acquire SIV.[146] However, SIV is a weak virus, and it is typically suppressed by the human immune system within weeks of infection. It is thought that several transmissions of the virus from individual to individual in quick succession are necessary to allow it enough time to mutate into HIV.[147] Furthermore, due to its relatively low person-to-person transmission rate, it can only spread throughout the population in the presence of one or more high-risk transmission channels, which are thought to have been absent in Africa prior to the 20th century.
Specific proposed high-risk transmission channels, allowing the virus to adapt to humans and spread throughout the society, depend on the proposed timing of the animal-to-human crossing. Genetic studies of the virus suggest that the most recent common ancestor of the HIV-1 M group dates back to circa 1910.[148] Proponents of this dating link the HIV epidemic with the emergence of colonialism and growth of large colonial African cities, leading to social changes, including different patterns of sexual contact (especially multiple, concurrent partnerships), the spread of prostitution, and the concomitant high frequency of genital ulcer diseases (such as syphilis) in nascent colonial cities.[149] While transmission rates of HIV during vaginal intercourse are typically low, they are increased manyfold if one of the partners suffers from a sexually transmitted infection resulting in genital ulcers. Early 1900s colonial cities were notable due to their high prevalence of prostitution and genital ulcers to the degree that as of 1928 as many as 45% of female residents of eastern Leopoldville were thought to have been prostitutes and as of 1933 around 15% of all residents of the same city were infected by one of the forms of syphilis.[149]
An alternative view—unsupported by evidence—holds that unsafe medical practices in Africa during years following World War II, such as unsterile reuse of single-use syringes during mass vaccination, antibiotic, and anti-malaria treatment campaigns, were the initial vector that allowed the virus to adapt to humans and spread.[147][150][151]
The earliest, well-documented case of HIV in a human dates back to 1959 in the Belgian Congo.[152] The virus may have been present in the United States as early as the mid-to-late 1950s, as a sixteen-year-old male presented with symptoms in 1966 and died in 1969.[153]
See also
- Antiviral drug
- Discovery and development of HIV-protease inhibitors
- HIV/AIDS denialism
- World AIDS Day
References
^ Weiss RA (May 1993). "How does HIV cause AIDS?". Science. 260 (5112): 1273–9. Bibcode:1993Sci...260.1273W. doi:10.1126/science.8493571. PMID 8493571..mw-parser-output cite.citation{font-style:inherit}.mw-parser-output q{quotes:"""""""'""'"}.mw-parser-output code.cs1-code{color:inherit;background:inherit;border:inherit;padding:inherit}.mw-parser-output .cs1-lock-free a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/6/65/Lock-green.svg/9px-Lock-green.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output .cs1-lock-limited a,.mw-parser-output .cs1-lock-registration a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/d/d6/Lock-gray-alt-2.svg/9px-Lock-gray-alt-2.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output .cs1-lock-subscription a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/a/aa/Lock-red-alt-2.svg/9px-Lock-red-alt-2.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output .cs1-subscription,.mw-parser-output .cs1-registration{color:#555}.mw-parser-output .cs1-subscription span,.mw-parser-output .cs1-registration span{border-bottom:1px dotted;cursor:help}.mw-parser-output .cs1-hidden-error{display:none;font-size:100%}.mw-parser-output .cs1-visible-error{font-size:100%}.mw-parser-output .cs1-subscription,.mw-parser-output .cs1-registration,.mw-parser-output .cs1-format{font-size:95%}.mw-parser-output .cs1-kern-left,.mw-parser-output .cs1-kern-wl-left{padding-left:0.2em}.mw-parser-output .cs1-kern-right,.mw-parser-output .cs1-kern-wl-right{padding-right:0.2em}
^ Douek DC, Roederer M, Koup RA (2009). "Emerging Concepts in the Immunopathogenesis of AIDS". Annual Review of Medicine. 60: 471–84. doi:10.1146/annurev.med.60.041807.123549. PMC 2716400. PMID 18947296.
^ UNAIDS, WHO (December 2007). "2007 AIDS epidemic update" (PDF). p. 10. Retrieved 2008-03-12.
^ Mabuka J, Nduati R, Odem-Davis K, Peterson D, Overbaugh J (2012). Desrosiers RC, ed. "HIV-Specific Antibodies Capable of ADCC Are Common in Breastmilk and Are Associated with Reduced Risk of Transmission in Women with High Viral Loads". PLOS Pathogens. 8 (6): e1002739. doi:10.1371/journal.ppat.1002739. PMC 3375288. PMID 22719248.
^ Hahn, Robert A.; Inhorn, Marcia Claire, eds. (2009). Anthropology and public health : bridging differences in culture and society (2nd ed.). Oxford: Oxford University Press. p. 449. ISBN 978-0-19-537464-3. OCLC 192042314.
^ Mead MN (2008). "Contaminants in human milk: weighing the risks against the benefits of breastfeeding". Environmental Health Perspectives. 116 (10): A426–34. doi:10.1289/ehp.116-a426. PMC 2569122. PMID 18941560. Archived from the original on 6 November 2008.
^ "Preventing Mother-to-Child Transmission of HIV". HIV.gov. Retrieved 2017-12-08.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain.
^ Cunningham AL, Donaghy H, Harman AN, Kim M, Turville SG (2010). "Manipulation of dendritic cell function by viruses". Current Opinion in Microbiology. 13 (4): 524–529. doi:10.1016/j.mib.2010.06.002. PMID 20598938.
^ Doitsh, Gilad; Galloway, Nicole L. K.; Geng, Xin; Yang, Zhiyuan; Monroe, Kathryn M.; Zepeda, Orlando; Hunt, Peter W.; Hatano, Hiroyu; Sowinski, Stefanie; Muñoz-Arias, Isa; Greene, Warner C. (2014). "Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection". Nature. 505 (7484): 509–514. Bibcode:2014Natur.505..509D. doi:10.1038/nature12940. PMC 4047036. PMID 24356306.
^ Garg H, Mohl J, Joshi A (Nov 9, 2012). "HIV-1 induced bystander apoptosis". Viruses. 4 (11): 3020–43. doi:10.3390/v4113020. PMC 3509682. PMID 23202514.
^ Kumar, Vinay (2012). Robbins Basic Pathology (9th ed.). p. 147. ISBN 978-1-4557-3787-1.
^ International Committee on Taxonomy of Viruses (2002). "61.0.6. Lentivirus". National Institutes of Health. Retrieved February 28, 2006.
^ International Committee on Taxonomy of Viruses (2002). "61. Retroviridae". National Institutes of Health. Retrieved February 28, 2006.
^ Levy JA (1993). "HIV pathogenesis and long-term survival". AIDS. 7 (11): 1401–10. doi:10.1097/00002030-199311000-00001. PMID 8280406.
^ Smith JA, Daniel R (2006). "Following the path of the virus: the exploitation of host DNA repair mechanisms by retroviruses". ACS Chemical Biology. 1 (4): 217–26. doi:10.1021/cb600131q. PMID 17163676.
^ ab Siliciano, R. F.; Greene, W. C. (2011). "HIV Latency". Cold Spring Harbor Perspectives in Medicine. 1 (1): a007096. doi:10.1101/cshperspect.a007096. PMC 3234450. PMID 22229121.
^ Gilbert PB, McKeague IW, Eisen G, Mullins C, Guéye-NDiaye A, Mboup S, Kanki PJ (February 28, 2003). "Comparison of HIV-1 and HIV-2 infectivity from a prospective cohort study in Senegal". Statistics in Medicine. 22 (4): 573–593. doi:10.1002/sim.1342. PMID 12590415.
^ ab Reeves JD, Doms RW (2002). "Human Immunodeficiency Virus Type 2" (PDF). Journal of General Virology. 83 (Pt 6): 1253–65. doi:10.1099/0022-1317-83-6-1253. PMID 12029140.
^ McGovern SL, Caselli E, Grigorieff N, Shoichet BK (2002). "A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening". Journal of Medicinal Chemistry. 45 (8): 1712–22. doi:10.1021/jm010533y. PMID 11931626.
^ Compared with overview in: Fisher, Bruce; Harvey, Richard P.; Champe, Pamela C. (2007). Lippincott's Illustrated Reviews: Microbiology. Lippincott's Illustrated Reviews. Hagerstown, MD: Lippincott Williams & Wilkins. p. 3. ISBN 978-0-7817-8215-9.
^ abcdefg Various (2008). HIV Sequence Compendium 2008 Introduction (PDF). Retrieved March 31, 2009.
^ abc Chan DC, Fass D, Berger JM, Kim PS (1997). "Core structure of gp41 from the HIV envelope glycoprotein" (PDF). Cell. 89 (2): 263–73. doi:10.1016/S0092-8674(00)80205-6. PMID 9108481.
^ Klein, Joshua S.; Bjorkman, Pamela J.; Rall, Glenn F. (27 May 2010). "Few and Far Between: How HIV May Be Evading Antibody Avidity". PLOS Pathogens. 6 (5): e1000908. doi:10.1371/journal.ppat.1000908. PMC 2877745. PMID 20523901.
^ National Institute of Health (June 17, 1998). "Crystal structure of key HIV protein reveals new prevention, treatment targets" (Press release). Archived from the original on February 19, 2006. Retrieved September 14, 2006.
^ Behrens, Anna-Janina; Vasiljevic, Snezana; Pritchard, Laura K; Harvey, David J; Andev, Rajinder S; Krumm, Stefanie A; Struwe, Weston B; Cupo, Albert; Kumar, Abhinav; Zitzmann, Nicole; Seabright, Gemma E; Kramer, Holger B; Spencer, Daniel I.R; Royle, Louise; Lee, Jeong Hyun; Klasse, Per J; Burton, Dennis R; Wilson, Ian A; Ward, Andrew B; Sanders, Rogier W; Moore, John P; Doores, Katie J; Crispin, Max (2016). "Composition and Antigenic Effects of Individual Glycan Sites of a Trimeric HIV-1 Envelope Glycoprotein". Cell Reports. 14 (11): 2695–706. doi:10.1016/j.celrep.2016.02.058. PMC 4805854. PMID 26972002.
^ Pritchard, Laura K; Spencer, Daniel I.R; Royle, Louise; Bonomelli, Camille; Seabright, Gemma E; Behrens, Anna-Janina; Kulp, Daniel W; Menis, Sergey; Krumm, Stefanie A; Dunlop, D. Cameron; Crispin, Daniel J; Bowden, Thomas A; Scanlan, Christopher N; Ward, Andrew B; Schief, William R; Doores, Katie J; Crispin, Max (2015). "Glycan clustering stabilizes the mannose patch of HIV-1 and preserves vulnerability to broadly neutralizing antibodies". Nature Communications. 6: 7479. Bibcode:2015NatCo...6E7479P. doi:10.1038/ncomms8479. PMC 4500839. PMID 26105115.
^ Pritchard, Laura K; Harvey, David J; Bonomelli, Camille; Crispin, Max; Doores, Katie J (2015). "Cell- and Protein-Directed Glycosylation of Native Cleaved HIV-1 Envelope". Journal of Virology. 89 (17): 8932–44. doi:10.1128/JVI.01190-15. PMC 4524065. PMID 26085151.
^ Crispin, Max; Doores, Katie J (2015). "Targeting host-derived glycans on enveloped viruses for antibody-based vaccine design". Current Opinion in Virology. 11: 63–9. doi:10.1016/j.coviro.2015.02.002. PMC 4827424. PMID 25747313.
^ Julien, Jean-Philippe; Cupo, Albert; Sok, Devin; Stanfield, Robyn L.; Lyumkis, Dmitry; Deller, Marc C.; Klasse, Per-Johan; Burton, Dennis R.; Sanders, Rogier W. (2013-12-20). "Crystal structure of a soluble cleaved HIV-1 envelope trimer". Science. 342 (6165): 1477–1483. Bibcode:2013Sci...342.1477J. doi:10.1126/science.1245625. ISSN 1095-9203. PMC 3886632. PMID 24179159.
^ Lyumkis, Dmitry; Julien, Jean-Philippe; de Val, Natalia; Cupo, Albert; Potter, Clinton S.; Klasse, Per-Johan; Burton, Dennis R.; Sanders, Rogier W.; Moore, John P. (2013-12-20). "Cryo-EM structure of a fully glycosylated soluble cleaved HIV-1 envelope trimer". Science. 342 (6165): 1484–1490. Bibcode:2013Sci...342.1484L. doi:10.1126/science.1245627. ISSN 1095-9203. PMC 3954647. PMID 24179160.
^ Sanders, Rogier W.; Derking, Ronald; Cupo, Albert; Julien, Jean-Philippe; Yasmeen, Anila; de Val, Natalia; Kim, Helen J.; Blattner, Claudia; de la Peña, Alba Torrents (2013-09-01). "A next-generation cleaved, soluble HIV-1 Env trimer, BG505 SOSIP.664 gp140, expresses multiple epitopes for broadly neutralizing but not non-neutralizing antibodies". PLOS Pathogens. 9 (9): e1003618. doi:10.1371/journal.ppat.1003618. ISSN 1553-7374. PMC 3777863. PMID 24068931.
^ Pritchard, Laura K.; Vasiljevic, Snezana; Ozorowski, Gabriel; Seabright, Gemma E.; Cupo, Albert; Ringe, Rajesh; Kim, Helen J.; Sanders, Rogier W.; Doores, Katie J. (2015-06-16). "Structural Constraints Determine the Glycosylation of HIV-1 Envelope Trimers". Cell Reports. 11 (10): 1604–1613. doi:10.1016/j.celrep.2015.05.017. ISSN 2211-1247. PMC 4555872. PMID 26051934.
^ de Taeye, Steven W.; Ozorowski, Gabriel; Torrents de la Peña, Alba; Guttman, Miklos; Julien, Jean-Philippe; van den Kerkhof, Tom L. G. M.; Burger, Judith A.; Pritchard, Laura K.; Pugach, Pavel (2015-12-17). "Immunogenicity of Stabilized HIV-1 Envelope Trimers with Reduced Exposure of Non-neutralizing Epitopes". Cell. 163 (7): 1702–1715. doi:10.1016/j.cell.2015.11.056. ISSN 1097-4172. PMC 4732737. PMID 26687358.
^ Ouellet DL, Plante I, Landry P, Barat C, Janelle ME, Flamand L, Tremblay MJ, Provost P (April 2008). "Identification of functional microRNAs released through asymmetrical processing of HIV-1 TAR element". Nucleic Acids Research. 36 (7): 2353–65. doi:10.1093/nar/gkn076. PMC 2367715. PMID 18299284.
^ Klase Z, Winograd R, Davis J, Carpio L, Hildreth R, Heydarian M, Fu S, McCaffrey T, Meiri E, Ayash-Rashkovsky M, Gilad S, Bentwich Z, Kashanchi F (2009). "HIV-1 TAR miRNA protects against apoptosis by altering cellular gene expression". Retrovirology. 6 (1): 18. doi:10.1186/1742-4690-6-18. PMC 2654423. PMID 19220914.
^ Vasudevan AA, Smits SH, Höppner A, Häussinger D, Koenig BW, Münk C (Nov 2013). "Structural features of antiviral DNA cytidine deaminases" (Submitted manuscript). Biological Chemistry. 394 (11): 1357–70. doi:10.1515/hsz-2013-0165. PMID 23787464.
^ Garcia JV, Miller AD (April 1991). "Serine phosphorylation-independent downregulation of cell-surface CD4 by nef". Nature. 350 (6318): 508–11. Bibcode:1991Natur.350..508G. doi:10.1038/350508a0. PMID 2014052.
^ Schwartz O, Maréchal V, Le Gall S, Lemonnier F, Heard JM (March 1996). "Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein". Nature Medicine. 2 (3): 338–42. doi:10.1038/nm0396-338. PMID 8612235.
^ Stumptner-Cuvelette P, Morchoisne S, Dugast M, Le Gall S, Raposo G, Schwartz O, Benaroch P (October 2001). "HIV-1 Nef impairs MHC class II antigen presentation and surface expression". Proceedings of the National Academy of Sciences of the United States of America. 98 (21): 12144–9. Bibcode:2001PNAS...9812144S. doi:10.1073/pnas.221256498. PMC 59782. PMID 11593029.
^ Arrildt, Kathryn Twigg; Joseph, Sarah Beth; Swanstrom, Ronald (March 2012). "The HIV-1 Env Protein: A Coat of Many Colors". Current HIV/AIDS Reports (Current HIV/AIDS Reports): 53–63.
^ ab Berger EA, Doms RW, Fenyö EM, Korber BT, Littman DR, Moore JP, Sattentau QJ, Schuitemaker H, Sodroski J, Weiss RA (1998). "A new classification for HIV-1". Nature. 391 (6664): 240. Bibcode:1998Natur.391..240B. doi:10.1038/34571. PMID 9440686.
^ abc Coakley E, Petropoulos CJ, Whitcomb JM (2005). "Assessing ch vbgemokine co-receptor usage in HIV". Current Opinion in Infectious Diseases. 18 (1): 9–15. doi:10.1097/00001432-200502000-00003. PMID 15647694.
^
Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, Davis CB, Peiper SC, Schall TJ, Littman DR, Landau NR (1996). "Identification of a major co-receptor for primary isolates of HIV-1". Nature. 381 (6584): 661–6. Bibcode:1996Natur.381..661D. doi:10.1038/381661a0. PMID 8649511.
^
Feng Y, Broder CC, Kennedy PE, Berger EA (1996). "HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor". Science. 272 (5263): 872–7. Bibcode:1996Sci...272..872F. doi:10.1126/science.272.5263.872. PMC 3412311. PMID 8629022.
^ Knight SC, Macatonia SE, Patterson S (1990). "HIV I infection of dendritic cells". International Review of Immunology. 6 (2–3): 163–75. doi:10.3109/08830189009056627. PMID 2152500.
^ Tang J, Kaslow RA (2003). "The impact of host genetics on HIV infection and disease progression in the era of highly active antiretroviral therapy". AIDS. 17 (Suppl 4): S51–S60. doi:10.1097/00002030-200317004-00006. PMID 15080180.
^ Zhu T, Mo H, Wang N, Nam DS, Cao Y, Koup RA, Ho DD (1993). "Genotypic and phenotypic characterization of HIV-1 patients with primary infection". Science. 261 (5125): 1179–81. Bibcode:1993Sci...261.1179Z. doi:10.1126/science.8356453. PMID 8356453.
^ van't Wout AB, Kootstra NA, Mulder-Kampinga GA, Albrecht-van Lent N, Scherpbier HJ, Veenstra J, Boer K, Coutinho RA, Miedema F, Schuitemaker H (1994). "Macrophage-tropic variants initiate human immunodeficiency virus type 1 infection after sexual, parenteral, and vertical transmission". Journal of Clinical Investigation. 94 (5): 2060–7. doi:10.1172/JCI117560. PMC 294642. PMID 7962552.
^ Zhu T, Wang N, Carr A, Nam DS, Moor-Jankowski R, Cooper DA, Ho DD (1996). "Genetic characterization of human immunodeficiency virus type 1 in blood and genital secretions: evidence for viral compartmentalization and selection during sexual transmission". Journal of Virology. 70 (5): 3098–107. PMC 190172. PMID 8627789.
^ Clevestig P, Maljkovic I, Casper C, Carlenor E, Lindgren S, Navér L, Bohlin AB, Fenyö EM, Leitner T, Ehrnst A (2005). "The X4 phenotype of HIV type 1 evolves from R5 in two children of mothers, carrying X4, and is not linked to transmission". AIDS Research and Human Retroviruses. 21 (5): 371–8. doi:10.1089/aid.2005.21.371. PMID 15929699.
^ Moore JP (1997). "Coreceptors: implications for HIV pathogenesis and therapy". Science. 276 (5309): 51–2. doi:10.1126/science.276.5309.51. PMID 9122710.
^ Karlsson A, Parsmyr K, Aperia K, Sandström E, Fenyö EM, Albert J (1994). "MT-2 cell tropism of human immunodeficiency virus type 1 isolates as a marker for response to treatment and development of drug resistance". The Journal of Infectious Diseases. 170 (6): 1367–75. doi:10.1093/infdis/170.6.1367. PMID 7995974.
^ Koot M, van 't Wout AB, Kootstra NA, de Goede RE, Tersmette M, Schuitemaker H (1996). "Relation between changes in cellular load, evolution of viral phenotype, and the clonal composition of virus populations in the course of human immunodeficiency virus type 1 infection". The Journal of Infectious Diseases. 173 (2): 349–54. doi:10.1093/infdis/173.2.349. PMID 8568295.
^ Cheney K, McKnight A (2010). "HIV-2 Tropism and Disease". Lentiviruses and Macrophages: Molecular and Cellular Interactions. Caister Academic Press. ISBN 978-1-904455-60-8.
[page needed]
^ abcdefg Chan DC, Kim PS (1998). "HIV entry and its inhibition". Cell. 93 (5): 681–4. doi:10.1016/S0092-8674(00)81430-0. PMID 9630213.
^ abcdef Wyatt R, Sodroski J (1998). "The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens". Science. 280 (5371): 1884–8. Bibcode:1998Sci...280.1884W. doi:10.1126/science.280.5371.1884. PMID 9632381.
^ ab Arthos J, Cicala C, Martinelli E, Macleod K, Van Ryk D, Wei D, Xiao Z, Veenstra TD, Conrad TP, Lempicki RA, McLaughlin S, Pascuccio M, Gopaul R, McNally J, Cruz CC, Censoplano N, Chung E, Reitano KN, Kottilil S, Goode DJ, Fauci AS (2008). "HIV-1 envelope protein binds to and signals through integrin alpha(4)beta(7), the gut mucosal homing receptor for peripheral T cells". Nature Immunology. 9 (3): 301–9. doi:10.1038/ni1566. PMID 18264102.
^ ab Pope M, Haase AT (2003). "Transmission, acute HIV-1 infection and the quest for strategies to prevent infection". Nature Medicine. 9 (7): 847–52. doi:10.1038/nm0703-847. PMID 12835704.
^ Haedicke J, Brown C, Naghavi MH (Aug 2009). "The brain-specific factor FEZ1 is a determinant of neuronal susceptibility to HIV-1 infection". Proceedings of the National Academy of Sciences. 106 (33): 14040–14045. Bibcode:2009PNAS..10614040H. doi:10.1073/pnas.0900502106. PMC 2729016. PMID 19667186.
^ Daecke J, Fackler OT, Dittmar MT, Kräusslich HG (2005). "Involvement of clathrin-mediated endocytosis in human immunodeficiency virus type 1 entry". Journal of Virology. 79 (3): 1581–1594. doi:10.1128/jvi.79.3.1581-1594.2005. PMC 544101. PMID 15650184.
^ Miyauchi K, Kim Y, Latinovic O, Morozov V, Melikyan GB (2009). "HIV Enters Cells via Endocytosis and Dynamin-Dependent Fusion with Endosomes". Cell. 137 (3): 433–444. doi:10.1016/j.cell.2009.02.046. PMC 2696170. PMID 19410541.
^ Koch P, Lampe M, Godinez WJ, Müller B, Rohr K, Kräusslich HG, Lehmann MJ (2009). "Visualizing fusion of pseudotyped HIV-1 particles in real time by live cell microscopy". Retrovirology. 6: 84. doi:10.1186/1742-4690-6-84. PMC 2762461. PMID 19765276.
^ Thorley JA, McKeating JA, Rappoport JZ (2010). "Mechanis ms of viral entry: sneaking in the front door". Protoplasma. 244 (1–4): 15–24. doi:10.1007/s00709-010-0152-6. PMC 3038234. PMID 20446005.
^ Permanyer M, Ballana E, Esté JA (2010). "Endocytosis of HIV: anything goes". Trends in Microbiology. 18 (12): 543–551. doi:10.1016/j.tim.2010.09.003. PMID 20965729.
^ abc Zheng YH, Lovsin N, Peterlin BM (2005). "Newly identified host factors modulate HIV replication". Immunology Letters. 97 (2): 225–34. doi:10.1016/j.imlet.2004.11.026. PMID 15752562.
^ "IV. Viruses> F. Animal Virus Life Cycles > 3. The Life Cycle of HIV". Doc Kaiser's Microbiology Home Page. Community College of Baltimore County. January 2008. Archived from the original on July 26, 2010.
^ Hiscott J, Kwon H, Génin P (2001). "Hostile takeovers: viral appropriation of the NF-kB pathway". Journal of Clinical Investigation. 107 (2): 143–151. doi:10.1172/JCI11918. PMC 199181. PMID 11160127.
^ Pollard VW, Malim MH (1998). "The HIV-1 Rev protein". Annual Review of Microbiology. 52: 491–532. doi:10.1146/annurev.micro.52.1.491. PMID 9891806.
^ Butsch, M.; Boris-Lawrie, K. (2002). "Destiny of Unspliced Retroviral RNA: Ribosome and/or Virion?". Journal of Virology. 76 (7): 3089–94. doi:10.1128/JVI.76.7.3089-3094.2002. PMC 136024. PMID 11884533.
^ Hellmund, Chris; Lever, Andrew M. L. (2016-07-14). "Coordination of Genomic RNA Packaging with Viral Assembly in HIV-1". Viruses. 8 (7): 192. doi:10.3390/v8070192. ISSN 1999-4915. PMC 4974527. PMID 27428992.
^ Ricci, E. P.; Herbreteau, C. H.; Decimo, D.; Schaupp, A.; Datta, S. A. K.; Rein, A.; Darlix, J. -L.; Ohlmann, T. (2008). "In vitro expression of the HIV-2 genomic RNA is controlled by three distinct internal ribosome entry segments that are regulated by the HIV protease and the Gag polyprotein". RNA. 14 (7): 1443–55. doi:10.1261/rna.813608. PMC 2441975. PMID 18495939.
^ ab Hu WS, Temin HM (1990). "Retroviral recombination and reverse transcription". Science. 250 (4985): 1227–33. Bibcode:1990Sci...250.1227H. doi:10.1126/science.1700865. PMID 1700865.
^ ab Charpentier C, Nora T, Tenaillon O, Clavel F, Hance AJ (2006). "Extensive recombination among human immunodeficiency virus type 1 quasispecies makes an important contribution to viral diversity in individual patients". Journal of Virology. 80 (5): 2472–82. doi:10.1128/JVI.80.5.2472-2482.2006. PMC 1395372. PMID 16474154.
^ Nora T, Charpentier C, Tenaillon O, Hoede C, Clavel F, Hance AJ (2007). "Contribution of recombination to the evolution of human immunodeficiency viruses expressing resistance to antiretroviral treatment". Journal of Virology. 81 (14): 7620–8. doi:10.1128/JVI.00083-07. PMC 1933369. PMID 17494080.
^ Chen J, Powell D, Hu WS (2006). "High frequency of genetic recombination is a common feature of primate lentivirus replication". Journal of Virology. 80 (19): 9651–8. doi:10.1128/JVI.00936-06. PMC 1617242. PMID 16973569.
^ ab Bonhoeffer S, Chappey C, Parkin NT, Whitcomb JM, Petropoulos CJ (2004). "Evidence for positive epistasis in HIV-1". Science. 306 (5701): 1547–50. Bibcode:2004Sci...306.1547B. doi:10.1126/science.1101786. PMID 15567861.
^ Israël N, Gougerot-Pocidalo MA (1997). "Oxidative stress in human immunodeficiency virus infection". Cellular and Molecular Life Sciences. 53 (11–12): 864–70. doi:10.1007/s000180050106. PMID 9447238.
^ Michod RE, Bernstein H, Nedelcu AM (May 2008). "Adaptive value of sex in microbial pathogens" (PDF). Infection, Genetics and Evolution. 8 (3): 267–85. doi:10.1016/j.meegid.2008.01.002. PMID 18295550.
^ Hallenberger S, Bosch V, Angliker H, Shaw E, Klenk HD, Garten W (November 26, 1992). "Inhibition of furin-mediated cleavage activation of HIV-1 glycoprotein gp160". Nature. 360 (6402): 358–61. Bibcode:1992Natur.360..358H. doi:10.1038/360358a0. PMID 1360148.
^ Gelderblom HR (1997). "Fine structure of HIV and SIV" (PDF). In Los Alamos National Laboratory. HIV sequence compendium. Los Alamos National Laboratory. pp. 31–44.
^ abcd Zhang C, Zhou S, Groppelli E, Pellegrino P, Williams I, Borrow P, Chain BM, Jolly C (2015). "Hybrid Spreading Mechanisms and T Cell Activation Shape the Dynamics of HIV-1 Infection". PLOS Computational Biology. 11 (4): e1004179. arXiv:1503.08992. Bibcode:2015PLSCB..11E4179Z. doi:10.1371/journal.pcbi.1004179. PMC 4383537. PMID 25837979.
^ ab Jolly C, Kashefi K, Hollinshead M, Sattentau QJ (2004). "HIV-1 cell to cell transfer across an Env-induced, actin-dependent synapse". Journal of Experimental Medicine. 199 (2): 283–293. doi:10.1084/jem.20030648. PMC 2211771. PMID 14734528.
^ Sattentau Q (2008). "Avoiding the void: cell-to-cell spread of human viruses". Nature Reviews Microbiology. 6 (11): 815–826. doi:10.1038/nrmicro1972. PMID 18923409.
^ Duncan CJ, Russell RA, Sattentau QJ (2013). "High multiplicity HIV-1 cell-to-cell transmission from macrophages to CD4+ T cells limits antiretroviral efficacy". AIDS. 27 (14): 2201–2206. doi:10.1097/QAD.0b013e3283632ec4. PMC 4714465. PMID 24005480.
^ Sewald X, Gonzalez DG, Haberman AM, Mothes W (2012). "In vivo imaging of virological synapses". Nature Communications. 3: 1320. Bibcode:2012NatCo...3E1320S. doi:10.1038/ncomms2338. PMC 3784984. PMID 23271654.
^ Sigal A, Kim JT, Balazs AB, Dekel E, Mayo A, Milo R, Baltimore D (2011). "Cell-to-cell spread of HIV permits ongoing replication despite antiretroviral therapy". Nature. 477 (7362): 95–98. Bibcode:2011Natur.477...95S. doi:10.1038/nature10347. PMID 21849975.
^ abcd Robertson DL, Hahn BH, Sharp PM (1995). "Recombination in AIDS viruses". Journal of Molecular Evolution. 40 (3): 249–59. Bibcode:1995JMolE..40..249R. doi:10.1007/BF00163230. PMID 7723052.
^ Rambaut A, Posada D, Crandall KA, Holmes EC (January 2004). "The causes and consequences of HIV evolution". Nature Reviews Genetics. 5 (52–61): 52–61. doi:10.1038/nrg1246. PMID 14708016.
^ Perelson AS, Ribeiro RM (October 2008). "Estimating drug efficacy and viral dynamic parameters: HIV and HCV" (Submitted manuscript). Statistics in Medicine. 27 (23): 4647–57. doi:10.1002/sim.3116. PMID 17960579.
^ ab Sodora DL, Allan JS, Apetrei C, Brenchley JM, Douek DC, Else JG, Estes JD, Hahn BH, Hirsch VM, Kaur A, Kirchhoff F, Muller-Trutwin M, Pandrea I, Schmitz JE, Silvestri G (2009). "Toward an AIDS vaccine: lessons from natural simian immunodeficiency virus infections of African nonhuman primate hosts". Nature Medicine. 15 (8): 861–865. doi:10.1038/nm.2013. PMC 2782707. PMID 19661993.
^ Holzammer S, Holznagel E, Kaul A, Kurth R, Norley S (2001). "High virus loads in naturally and experimentally SIVagm-infected African green monkeys". Virology. 283 (2): 324–31. doi:10.1006/viro.2001.0870. PMID 11336557.
^ Kurth, R.; Norley, S. (1996). "Why don't the natural hosts of SIV develop simian AIDS?". The Journal of NIH Research. 8: 33–37.
^ Baier M, Dittmar MT, Cichutek K, Kurth R (1991). "Development of vivo of genetic variability of simian immunodeficiency virus". Proceedings of the National Academy of Sciences of the United States of America. 88 (18): 8126–30. Bibcode:1991PNAS...88.8126B. doi:10.1073/pnas.88.18.8126. PMC 52459. PMID 1896460.
^ Daniel MD, King NW, Letvin NL, Hunt RD, Sehgal PK, Desrosiers RC (1984). "A new type D retrovirus isolated from macaques with an immunodeficiency syndrome". Science. 223 (4636): 602–5. Bibcode:1984Sci...223..602D. doi:10.1126/science.6695172. PMID 6695172.
^ ab Keele BF, Jones JH, Terio KA, Estes JD, Rudicell RS, Wilson ML, Li Y, Learn GH, Beasley TM, Schumacher-Stankey J, Wroblewski E, Mosser A, Raphael J, Kamenya S, Lonsdorf EV, Travis DA, Mlengeya T, Kinsel MJ, Else JG, Silvestri G, Goodall J, Sharp PM, Shaw GM, Pusey AE, Hahn BH (2009). "Increased mortality and AIDS-like immunopathology in wild chimpanzees infected with SIVcpz". Nature. 460 (7254): 515–519. Bibcode:2009Natur.460..515K. doi:10.1038/nature08200. PMC 2872475. PMID 19626114.
^ Schindler M, Münch J, Kutsch O, Li H, Santiago ML, Bibollet-Ruche F, Müller-Trutwin MC, Novembre FJ, Peeters M, Courgnaud V, Bailes E, Roques P, Sodora DL, Silvestri G, Sharp PM, Hahn BH, Kirchhoff F (2006). "Nef-mediated suppression of T cell activation was lost in a lentiviral lineage that gave rise to HIV-1". Cell. 125 (6): 1055–67. doi:10.1016/j.cell.2006.04.033. PMID 16777597.
^ Thomson MM, Pérez-Alvarez L, Nájera R (2002). "Molecular epidemiology of HIV-1 genetic forms and its significance for vaccine development and therapy". The Lancet Infectious Diseases. 2 (8): 461–471. doi:10.1016/S1473-3099(02)00343-2. PMID 12150845.
^ Carr JK, Foley BT, Leitner T, Salminen M, Korber B, McCutchan F (1998). "Reference sequences representing the principal genetic diversity of HIV-1 in the pandemic" (PDF). In Los Alamos National Laboratory. HIV sequence compendium. Los Alamos, New Mexico: Los Alamos National Laboratory. pp. 10–19.CS1 maint: Uses authors parameter (link)
^ Osmanov S, Pattou C, Walker N, Schwardländer B, Esparza J (2002). "Estimated global distribution and regional spread of HIV-1 genetic subtypes in the year 2000". Acquired Immune Deficiency Syndrome. 29 (2): 184–190. doi:10.1097/00042560-200202010-00013. PMID 11832690.
^ Perrin L, Kaiser L, Yerly S (2003). "Travel and the spread of HIV-1 genetic variants". The Lancet Infectious Diseases. 3 (1): 22–27. doi:10.1016/S1473-3099(03)00484-5. PMID 12505029.
^ ab Plantier JC, Leoz M, Dickerson JE, De Oliveira F, Cordonnier F, Lemée V, Damond F, Robertson DL, Simon F (August 2009). "A new human immunodeficiency virus derived from gorillas". Nature Medicine. 15 (8): 871–2. doi:10.1038/nm.2016. PMID 19648927. Lay summary.
^ Keele BF, Van Heuverswyn F, Li Y, Bailes E, Takehisa J, Santiago ML, Bibollet-Ruche F, Chen Y, Wain LV, Liegeois F, Loul S, Ngole EM, Bienvenue Y, Delaporte E, Brookfield JF, Sharp PM, Shaw GM, Peeters M, Hahn BH (Jul 28, 2006). "Chimpanzee reservoirs of pandemic and nonpandemic HIV-1". Science. 313 (5786): 523–6. Bibcode:2006Sci...313..523K. doi:10.1126/science.1126531. PMC 2442710. PMID 16728595.
^ abcd
Kumaranayake, L.; Watts, C. (2001). "Resource allocation and priority setting of HIV/AIDS interventions: addressing the generalized epidemic in sub-Saharan Africa". Journal of International Development. 13 (4): 451–466. doi:10.1002/jid.797.
^ Kleinman S (September 2004). "Patient information: Blood donation and transfusion". Uptodate. Archived from the original on April 12, 2008.
^ ab Centers for Disease Control and Prevention (2001). "Revised guidelines for HIV counseling, testing, and referral". MMWR Recommendations and Reports. 50 (RR–19): 1–57. PMID 11718472.
^ Celum CL, Coombs RW, Lafferty W, Inui TS, Louie PH, Gates CA, McCreedy BJ, Egan R, Grove T, Alexander S (1991). "Indeterminate human immunodeficiency virus type 1 western blots: seroconversion risk, specificity of supplemental tests, and an algorithm for evaluation". The Journal of Infectious Diseases. 164 (4): 656–664. doi:10.1093/infdis/164.4.656. PMID 1894929.
^ "Country Comparison :: HIV/AIDS - Deaths". The World Factbook, Central Intelligence Agency.
^ Chou, Roger; Selph, Shelley; Dana, Tracy; Bougatsos, Christina; Zakher, Bernadette; Blazina, Ian; Korthuis, P. Todd (2012-11-20). "Screening for HIV: systematic review to update the 2005 U.S. Preventive Services Task Force recommendation". Annals of Internal Medicine. 157 (10): 706–718. doi:10.7326/0003-4819-157-10-201211200-00007. ISSN 1539-3704. PMID 23165662.
^ Chou R, Huffman LH, Fu R, Smits AK, Korthuis PT (July 2005). "Screening for HIV: a review of the evidence for the U.S. Preventive Services Task Force". Annals of Internal Medicine. 143 (1): 55–73. doi:10.7326/0003-4819-143-1-200507050-00010. PMID 15998755.
^ Tolle MA, Schwarzwald HL (July 15, 2010). "Postexposure prophylaxis against human immunodeficiency virus". American Family Physician. 82 (2): 161–6. PMID 20642270.
^ "Quick Reference Guide—Laboratory Testing for the Diagnosis of HIV Infection: Updated Recommendations" (PDF). cdc.gov. New York State Department of Health. June 27, 2014. pp. 1–2. Retrieved April 13, 2017.
^ "HIV Treatment: FDA-Approved HIV Medicines". AIDSinfo.
^ Bernstein, Ryan Lenora Brown, Lenny. "New HIV vaccine trial, the first in years, to begin". chicagotribune.com. Retrieved 2016-11-28.
^ Huang Y, Yu J, Lanzi A, Yao X, Andrews C, Tsai L, Gajjar M, Sun M, Seaman M, Padte N, Ho D (2016). "Engineered Bispecific Antibodies with Exquisite HIV-1-Neutralizing Activity". Cell. 165 (7): 1621–1631. doi:10.1016/j.cell.2016.05.024. PMC 4972332. PMID 27315479.
^ Bournazos, S., Gazumyan, A., Seaman, M., Nussenzweig, M. and Ravetch, J. (2016). Bispecific Anti-HIV-1 Antibodies with Enhanced Breadth and Potency.
^ Attia, Suzanna; Egger, Matthias; Müller, Monika; Zwahlen, Marcel; Low, Nicola (2009). "Sexual transmission of HIV according to viral load and antiretroviral therapy: Systematic review and meta-analysis". AIDS. 23 (11): 1397–404. doi:10.1097/QAD.0b013e32832b7dca. PMID 19381076.
^ Wilson, David P; Law, Matthew G; Grulich, Andrew E; Cooper, David A; Kaldor, John M (2008). "Relation between HIV viral load and infectiousness: A model-based analysis". The Lancet. 372 (9635): 314–20. doi:10.1016/S0140-6736(08)61115-0. PMID 18657710.
^ Cohen, Myron S; Chen, Ying Q; McCauley, Marybeth; Gamble, Theresa; Hosseinipour, Mina C; Kumarasamy, Nagalingeswaran; Hakim, James G; Kumwenda, Johnstone; Grinsztejn, Beatriz; Pilotto, Jose H.S; Godbole, Sheela V; Mehendale, Sanjay; Chariyalertsak, Suwat; Santos, Breno R; Mayer, Kenneth H; Hoffman, Irving F; Eshleman, Susan H; Piwowar-Manning, Estelle; Wang, Lei; Makhema, Joseph; Mills, Lisa A; De Bruyn, Guy; Sanne, Ian; Eron, Joseph; Gallant, Joel; Havlir, Diane; Swindells, Susan; Ribaudo, Heather; Elharrar, Vanessa; et al. (2011). "Prevention of HIV-1 Infection with Early Antiretroviral Therapy". New England Journal of Medicine. 365 (6): 493–505. doi:10.1056/NEJMoa1105243. PMC 3200068. PMID 21767103.
^ "HIV Consensus Statement". Prevention Access Campaign. November 19, 2017.
^ Mandell, Gerald L.; Bennett, John E.; Dolin, Raphael, eds. (2010). "Chapter 169". Mandell, Douglas, and Bennett's principles and practice of infectious diseases (7th ed.). Philadelphia: Churchill Livingstone/Elsevier. ISBN 978-0-443-06839-3.
[page needed]
^ Gottlieb MS (2006). "Pneumocystis pneumonia—Los Angeles. 1981". American Journal of Public Health. 96 (6): 980–1, discussion 982–3. doi:10.2105/AJPH.96.6.980. PMC 1470612. PMID 16714472. Archived from the original on April 22, 2009.
^ Friedman-Kien AE (October 1981). "Disseminated Kaposi's sarcoma syndrome in young homosexual men". Journal of the American Academy of Dermatology. 5 (4): 468–71. doi:10.1016/S0190-9622(81)80010-2. PMID 7287964.
^ Hymes KB, Cheung T, Greene JB, Prose NS, Marcus A, Ballard H, William DC, Laubenstein LJ (September 1981). "Kaposi's sarcoma in homosexual men-a report of eight cases". The Lancet. 2 (8247): 598–600. doi:10.1016/S0140-6736(81)92740-9. PMID 6116083.
^ ab Basavapathruni A, Anderson KS (December 2007). "Reverse transcription of the HIV-1 pandemic". The FASEB Journal. 21 (14): 3795–3808. doi:10.1096/fj.07-8697rev. PMID 17639073.
^ Lederberg, Joshua, ed. (2000). Encyclopedia of Microbiology (2nd ed.). Burlington: Elsevier. p. 106. ISBN 978-0-08-054848-7. Retrieved 9 June 2016.
^ Centers for Disease Control (1982). "Persistent, generalized lymphadenopathy among homosexual males". Morbidity and Mortality Weekly Report. 31 (19): 249–251. PMID 6808340.
^ Barré-Sinoussi F, Chermann JC, Rey F, Nugeyre MT, Chamaret S, Gruest J, Dauguet C, Axler-Blin C, Vézinet-Brun F, Rouzioux C, Rozenbaum W, Montagnier L (1983). "Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS)". Science. 220 (4599): 868–871. Bibcode:1983Sci...220..868B. doi:10.1126/science.6189183. PMID 6189183.
^ ab Centers for Disease Control (1982). "Opportunistic infections and Kaposi's sarcoma among Haitians in the United States". Morbidity and Mortality Weekly Report. 31 (26): 353–354, 360–361. PMID 6811853.
^ Altman LK (May 11, 1982). "New homosexual disorder worries health officials". The New York Times. Retrieved August 31, 2011.
^ Gilman, Sander L (1987). Gilman, Sander L., ed. "AIDS and Syphilis: The Iconography of Disease". October. 43: 87. doi:10.2307/3397566. JSTOR 3397566.
^ "Making Headway Under Hellacious Circumstances" (PDF). American Association for the Advancement of Science. July 28, 2006. Retrieved June 23, 2008.
^ Kher U (July 27, 1982). "A Name for the Plague". Time. Archived from the original on March 7, 2008. Retrieved March 10, 2008.
^ Centers for Disease Control (1982). "Update on acquired immune deficiency syndrome (AIDS)—United States". Morbidity and Mortality Weekly Report. 31 (37): 507–508, 513–514. PMID 6815471.
^ Gallo RC, Sarin PS, Gelmann EP, Robert-Guroff M, Richardson E, Kalyanaraman VS, Mann D, Sidhu GD, Stahl RE, Zolla-Pazner S, Leibowitch J, Popovic M (1983). "Isolation of human T-cell leukemia virus in acquired immune deficiency syndrome (AIDS)". Science. 220 (4599): 865–867. Bibcode:1983Sci...220..865G. doi:10.1126/science.6601823. PMID 6601823.
^ Barré-Sinoussi F, Chermann JC, Rey F, Nugeyre MT, Chamaret S, Gruest J, Dauguet C, Axler-Blin C, Vézinet-Brun F, Rouzioux C, Rozenbaum W, Montagnier L (1983). "Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS)". Science. 220 (4599): 868–871. Bibcode:1983Sci...220..868B. doi:10.1126/science.6189183. PMID 6189183.
^ "The 2008 Nobel Prize in Physiology or Medicine - Press Release". www.nobelprize.org. Retrieved 2018-01-28.
^ Aldrich, Robert; Wotherspoon, Garry, eds. (2001). Who's who in gay and lesbian history. London: Routledge. p. 154. ISBN 978-0-415-22974-6.
^ Levy JA; et al. (1984). "Isolation of lymphocytopathic retroviruses from San Francisco patients with AIDS". Science. 225 (4664): 840–842. Bibcode:1984Sci...225..840L. doi:10.1126/science.6206563.CS1 maint: Explicit use of et al. (link)
^ Levy JA, Kaminsky LS, Morrow WJW, Steimer K, Luciw P, Dina D, Hoxie J, Oshiro L (1985). "Infection by the retrovirus associated with the acquired immunodeficiency syndrome". Annals of Internal Medicine. 103 (5): 694–699. doi:10.7326/0003-4819-103-5-694.CS1 maint: Multiple names: authors list (link)
^ Sharp PM, Hahn BH (2011). "Origins of HIV and the AIDS Pandemic". Cold Spring Harbor Perspectives in Medicine. 1 (1): a006841. doi:10.1101/cshperspect.a006841. PMC 3234451. PMID 22229120.
^ Faria NR, Rambaut A, Suchard MA, Baele G, Bedford T, Ward MJ, Tatem AJ, Sousa JD, Arinaminpathy N, Pépin J, Posada D, Peeters M, Pybus OG, Lemey P (2014). "The early spread and epidemic ignition of HIV-1 in human populations". Science. 346 (6205): 56–61. Bibcode:2014Sci...346...56F. doi:10.1126/science.1256739. PMC 4254776. PMID 25278604.
^ Gao F, Bailes E, Robertson DL, Chen Y, Rodenburg CM, Michael SF, Cummins LB, Arthur LO, Peeters M, Shaw GM, Sharp PM, Hahn BH (1999). "Origin of HIV-1 in the chimpanzee Pan troglodytes troglodytes". Nature. 397 (6718): 436–41. Bibcode:1999Natur.397..436G. doi:10.1038/17130. PMID 9989410.
^ Keele BF, Van Heuverswyn F, Li Y, Bailes E, Takehisa J, Santiago ML, Bibollet-Ruche F, Chen Y, Wain LV, Liegeois F, Loul S, Ngole EM, Bienvenue Y, Delaporte E, Brookfield JF, Sharp PM, Shaw GM, Peeters M, Hahn BH (2006). "Chimpanzee reservoirs of pandemic and nonpandemic HIV-1". Science. 313 (5786): 523–6. Bibcode:2006Sci...313..523K. doi:10.1126/science.1126531. PMC 2442710. PMID 16728595.
^ Goodier JL, Kazazian HH (2008). "Retrotransposons revisited: the restraint and rehabilitation of parasites". Cell. 135 (1): 23–35. doi:10.1016/j.cell.2008.09.022. PMID 18854152.
^ Sharp PM, Bailes E, Chaudhuri RR, Rodenburg CM, Santiago MO, Hahn BH (2001). "The origins of acquired immune deficiency syndrome viruses: where and when?" (PDF). Philosophical Transactions of the Royal Society B. 356 (1410): 867–76. doi:10.1098/rstb.2001.0863. PMC 1088480. PMID 11405934.
^ Kalish ML, Wolfe ND, Ndongmo CB, McNicholl J, Robbins KE, Aidoo M, Fonjungo PN, Alemnji G, Zeh C, Djoko CF, Mpoudi-Ngole E, Burke DS, Folks TM (2005). "Central African hunters exposed to simian immunodeficiency virus". Emerging Infectious Diseases. 11 (12): 1928–30. doi:10.3201/eid1112.050394. PMC 3367631. PMID 16485481.
^ ab Marx PA, Alcabes PG, Drucker E (2001). "Serial human passage of simian immunodeficiency virus by unsterile injections and the emergence of epidemic human immunodeficiency virus in Africa" (PDF). Philosophical Transactions of the Royal Society B. 356 (1410): 911–20. doi:10.1098/rstb.2001.0867. PMC 1088484. PMID 11405938.
^ Worobey M, Gemmel M, Teuwen DE, Haselkorn T, Kunstman K, Bunce M, Muyembe JJ, Kabongo JM, Kalengayi RM, Van Marck E, Gilbert MT, Wolinsky SM (2008). "Direct evidence of extensive diversity of HIV-1 in Kinshasa by 1960". Nature. 455 (7213): 661–4. Bibcode:2008Natur.455..661W. doi:10.1038/nature07390. PMC 3682493. PMID 18833279.
^ ab de Sousa JD, Müller V, Lemey P, Vandamme AM (2010). Martin DP, ed. "High GUD incidence in the early 20th century created a particularly permissive time window for the origin and initial spread of epidemic HIV strains". PLOS One. 5 (4): e9936. Bibcode:2010PLoSO...5.9936S. doi:10.1371/journal.pone.0009936. PMC 2848574. PMID 20376191.
^ Chitnis A, Rawls D, Moore J (2000). "Origin of HIV type 1 in colonial French equatorial Africa?". AIDS Research and Human Retroviruses. 16 (1): 5–8. doi:10.1089/088922200309548. PMID 10628811.
^ Donald McNeil, Jr. (September 16, 2010). "Precursor to H.I.V. was in monkeys for millennia". The New York Times. Retrieved September 17, 2010.Dr. Marx believes that the crucial event was the introduction into Africa of millions of inexpensive, mass-produced syringes in the 1950s. ... suspect that the growth of colonial cities is to blame. Before 1910, no Central African town had more than 10,000 people. But urban migration rose, increasing sexual contacts and leading to red-light districts.
^ Zhu T, Korber BT, Nahmias AJ, Hooper E, Sharp PM, Ho DD (1998). "An African HIV-1 Sequence from 1959 and Implications for the Origin of the epidemic". Nature. 391 (6667): 594–7. Bibcode:1998Natur.391..594Z. doi:10.1038/35400. PMID 9468138.
^ Kolata, Gina (October 28, 1987). "Boy's 1969 death suggests AIDS invaded U.S. several times". The New York Times. Retrieved February 11, 2009.
Further reading
.mw-parser-output .refbegin{font-size:90%;margin-bottom:0.5em}.mw-parser-output .refbegin-hanging-indents>ul{list-style-type:none;margin-left:0}.mw-parser-output .refbegin-hanging-indents>ul>li,.mw-parser-output .refbegin-hanging-indents>dl>dd{margin-left:0;padding-left:3.2em;text-indent:-3.2em;list-style:none}.mw-parser-output .refbegin-100{font-size:100%}
Berlier W, Bourlet T, Lawrence P, Hamzeh H, Lambert C, Genin C, Verrier B, Dieu-Nosjean MC, Pozzetto B, Delézay O (2005). "Selective sequestration of X4 isolates by human genital epithelial cells: Implication for virus tropism selection process during sexual transmission of HIV". Journal of Medical Virology. 77 (4): 465–74. doi:10.1002/jmv.20478. PMID 16254974.
Joint United Nations Programme on HIV/AIDS (UNAIDS) (2011). Global HIV/AIDS Response, Epidemic update and health sector progress towards universal access (PDF). Joint United Nations Programme on HIV/AIDS.
Muciaccia B, Padula F, Vicini E, Gandini L, Lenzi A, Stefanini M (2005). "Beta-chemokine receptors 5 and 3 are expressed on the head region of human spermatozoon". The FASEB Journal. 19 (14): 2048–50. doi:10.1096/fj.05-3962fje. PMID 16174786.
External links
 Definitions from Wiktionary
Definitions from Wiktionary
 Media from Wikimedia Commons
Media from Wikimedia Commons
 News from Wikinews
News from Wikinews
 Textbooks from Wikibooks
Textbooks from Wikibooks
HIV/AIDS at Curlie
Authority control |
|
|---|
Categories:
- HIV/AIDS
- Causes of death
- Discovery and invention controversies
- IARC Group 2B carcinogens
- Lentiviruses
- Sexually transmitted diseases and infections
- 1983 in biology
(window.RLQ=window.RLQ||).push(function(){mw.config.set({"wgPageParseReport":{"limitreport":{"cputime":"2.344","walltime":"2.542","ppvisitednodes":{"value":13909,"limit":1000000},"ppgeneratednodes":{"value":0,"limit":1500000},"postexpandincludesize":{"value":539842,"limit":2097152},"templateargumentsize":{"value":8844,"limit":2097152},"expansiondepth":{"value":24,"limit":40},"expensivefunctioncount":{"value":24,"limit":500},"unstrip-depth":{"value":1,"limit":20},"unstrip-size":{"value":495118,"limit":5000000},"entityaccesscount":{"value":10,"limit":400},"timingprofile":["100.00% 2117.611 1 -total"," 66.59% 1410.184 1 Template:Reflist"," 50.56% 1070.635 126 Template:Cite_journal"," 6.27% 132.796 1 Template:Taxobox"," 5.73% 121.430 1 Template:Taxobox/core"," 4.61% 97.637 27 Template:Delink"," 4.07% 86.272 1 Template:Taxonbar"," 3.45% 73.001 20 Template:Taxobox_colour"," 3.01% 63.670 11 Template:Cite_book"," 2.78% 58.838 7 Template:Fix"]},"scribunto":{"limitreport-timeusage":{"value":"1.419","limit":"10.000"},"limitreport-memusage":{"value":8477059,"limit":52428800}},"cachereport":{"origin":"mw1262","timestamp":"20181101014607","ttl":1900800,"transientcontent":false}}});mw.config.set({"wgBackendResponseTime":137,"wgHostname":"mw1272"});});

 Clash Royale CLAN TAG
Clash Royale CLAN TAG

